
Comparative Genomic Analysis of Lactiplantibacillus plantarum: Insights into Its Genetic Diversity, Metabolic Function, and Antibiotic Resistance

Abstract
1. Introduction
2. Materials and Methods
2.1. Collection of Microbial Genomes
2.2. Construction of the Phylogenetic Tree
2.3. Prediction of Metabolic Profiles
2.4. In Silico Analysis of Virulence Genes and Antimicrobial Resistance and Their Associated Mobile Genetic Elements (MGEs)
2.5. In Silico Analysis of Antimicrobial Peptides (AMPs)
2.6. Statistics and Visualization
3. Results
3.1. Strain Information and Genome Characteristics
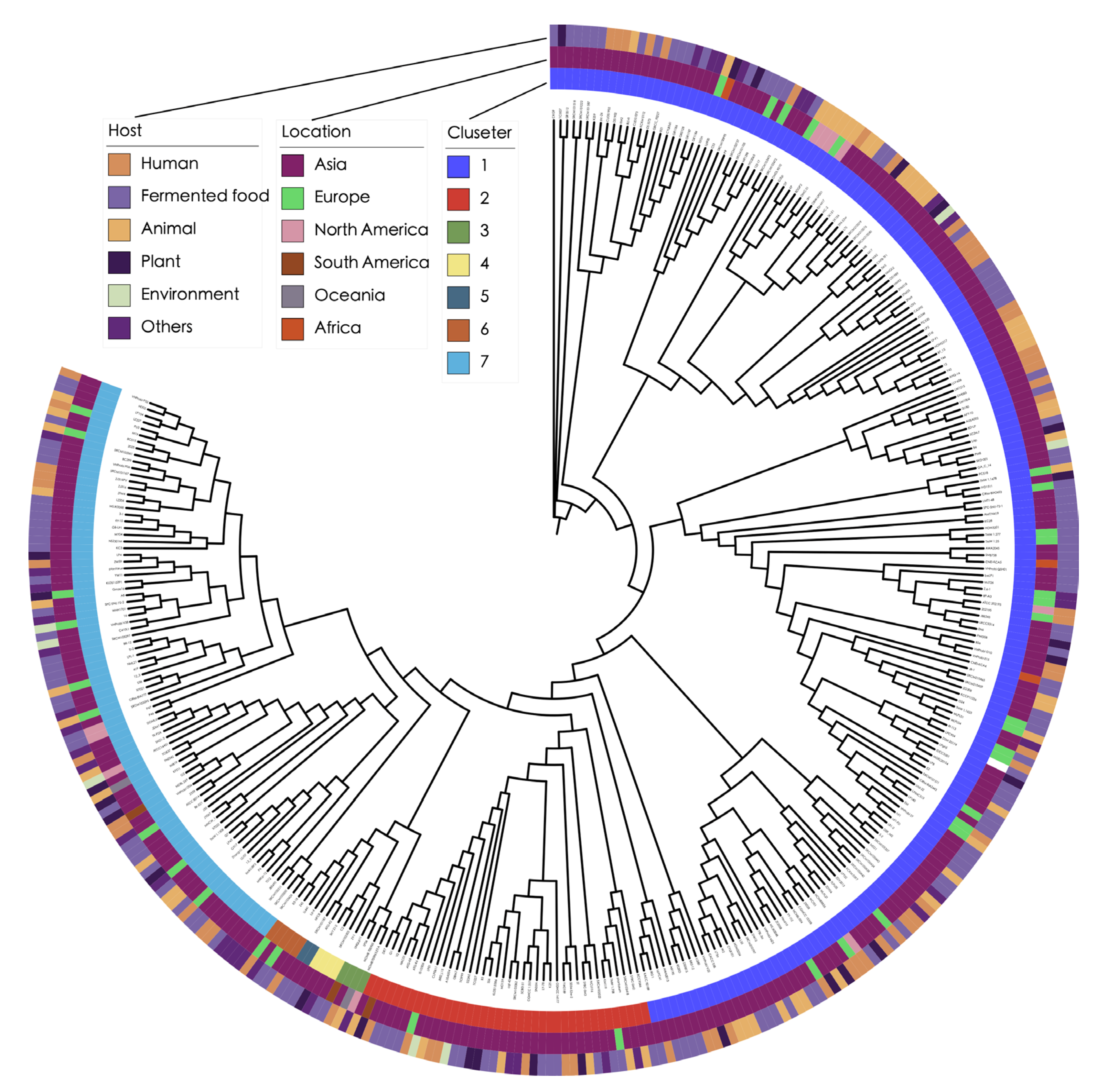
3.2. Phylogenetic Analysis Reveals Complex Transmission Pathways
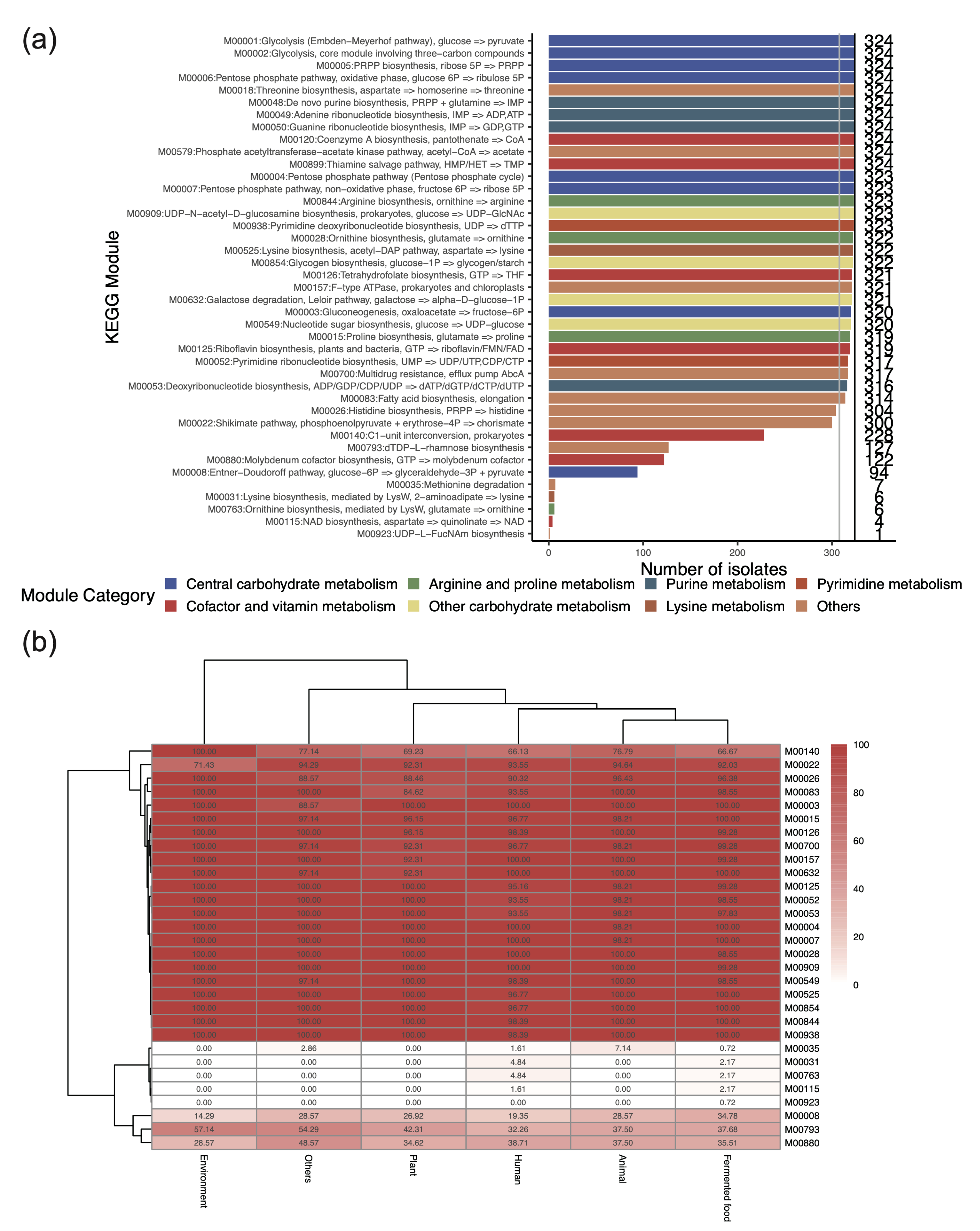
3.3. Key Metabolic Functions Revealed by the Presence of KEGG Modules in L. plantarum
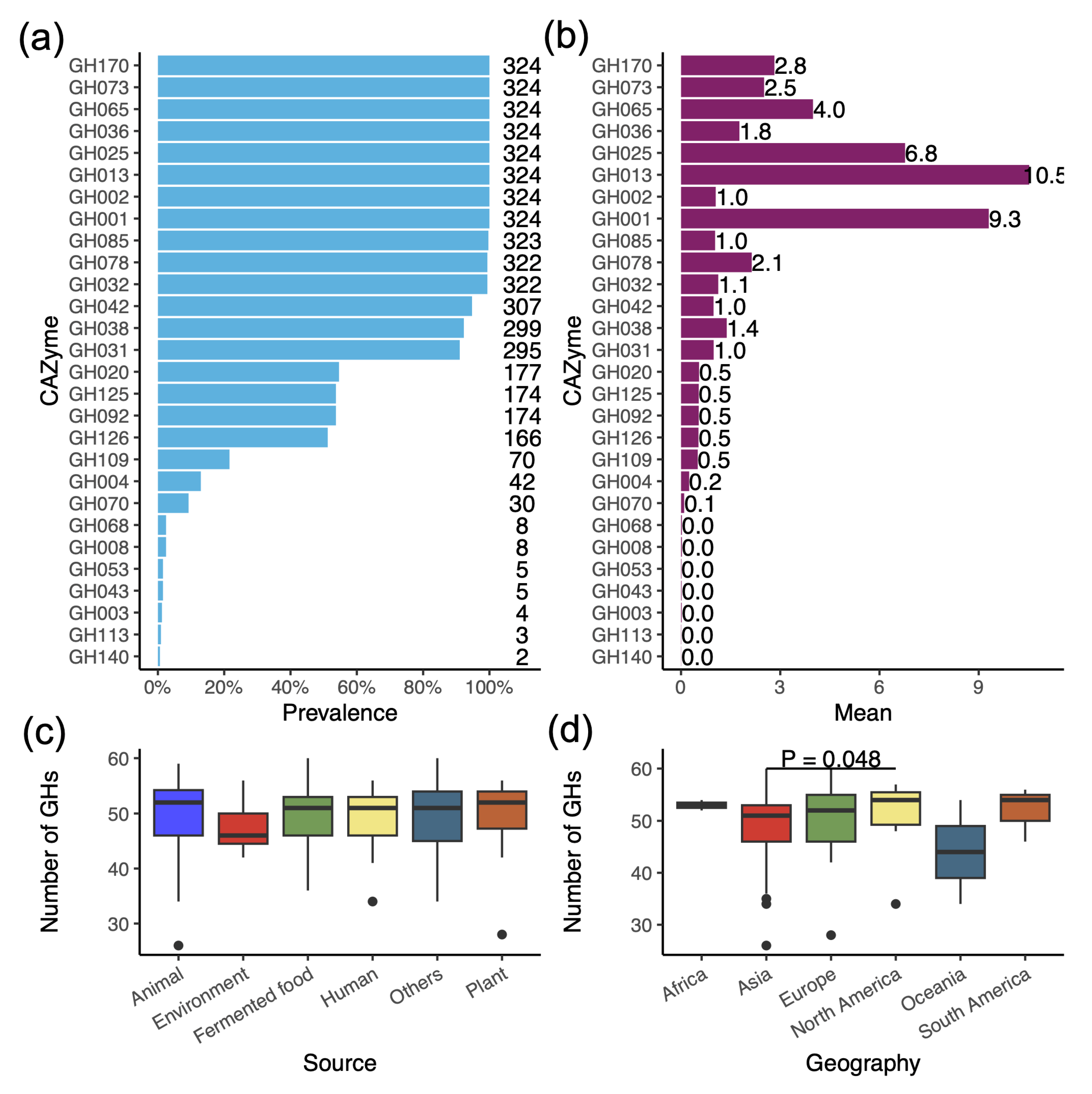
3.4. Profile of Glycoside Hydrolases (GHs) Reveals the Ability to Degrade a Variety of Carbohydrates
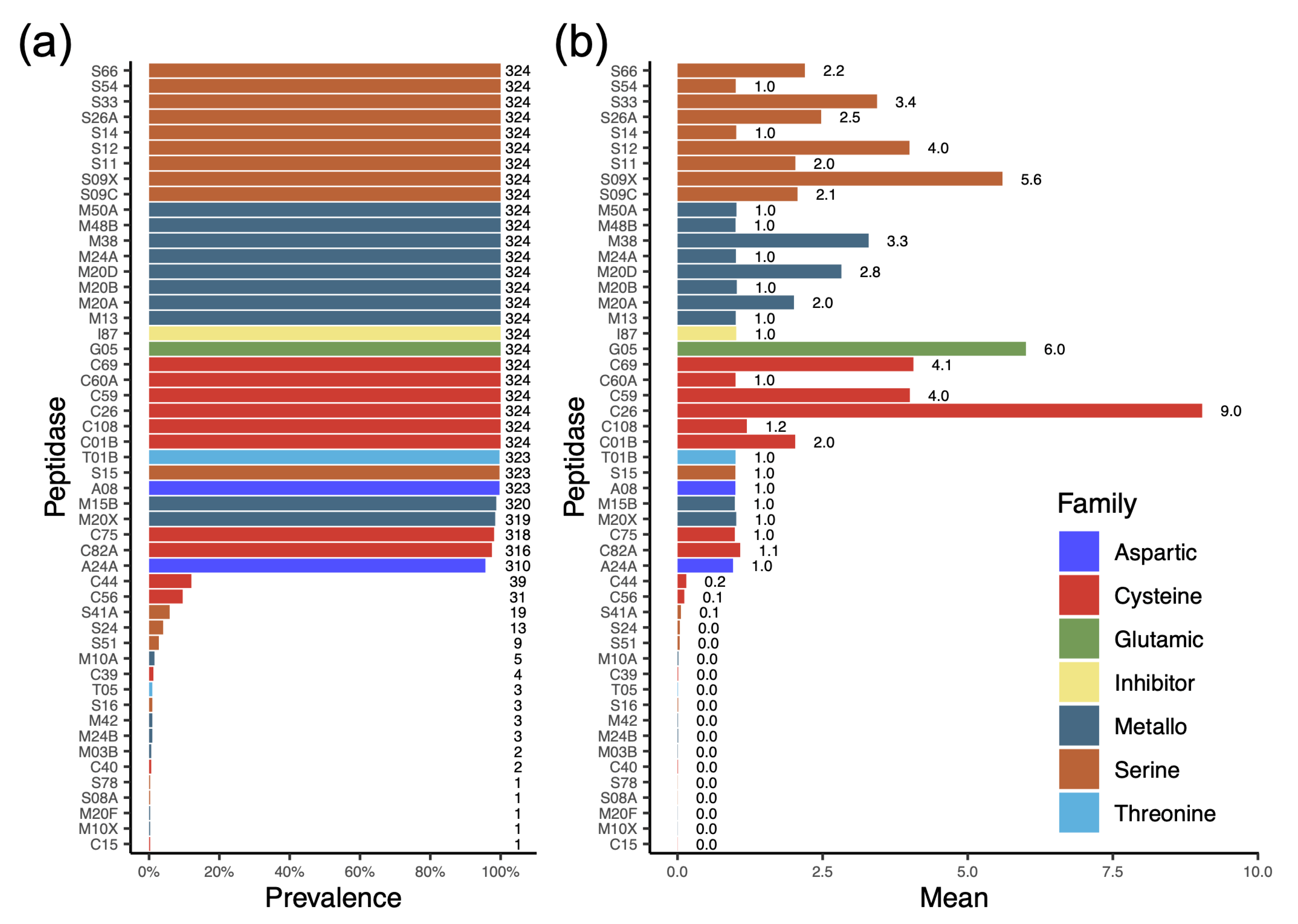
3.5. Profile of Protease Types Reveals Their Ability to Degrade or Modify a Variety of Proteins
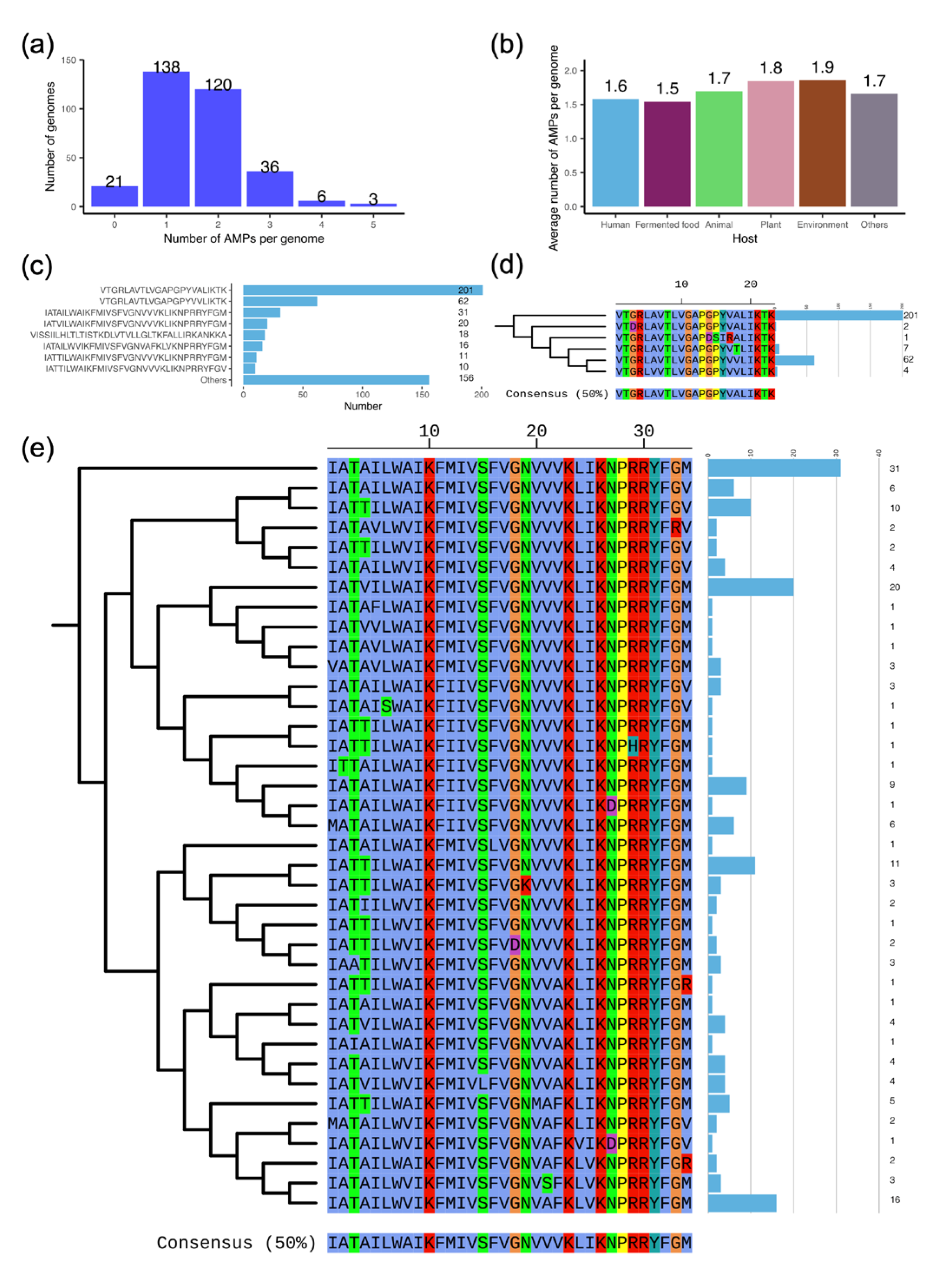
3.6. The Antimicrobial Peptide (AMP) Facilitates the Antimicrobial Properties of L. plantarum
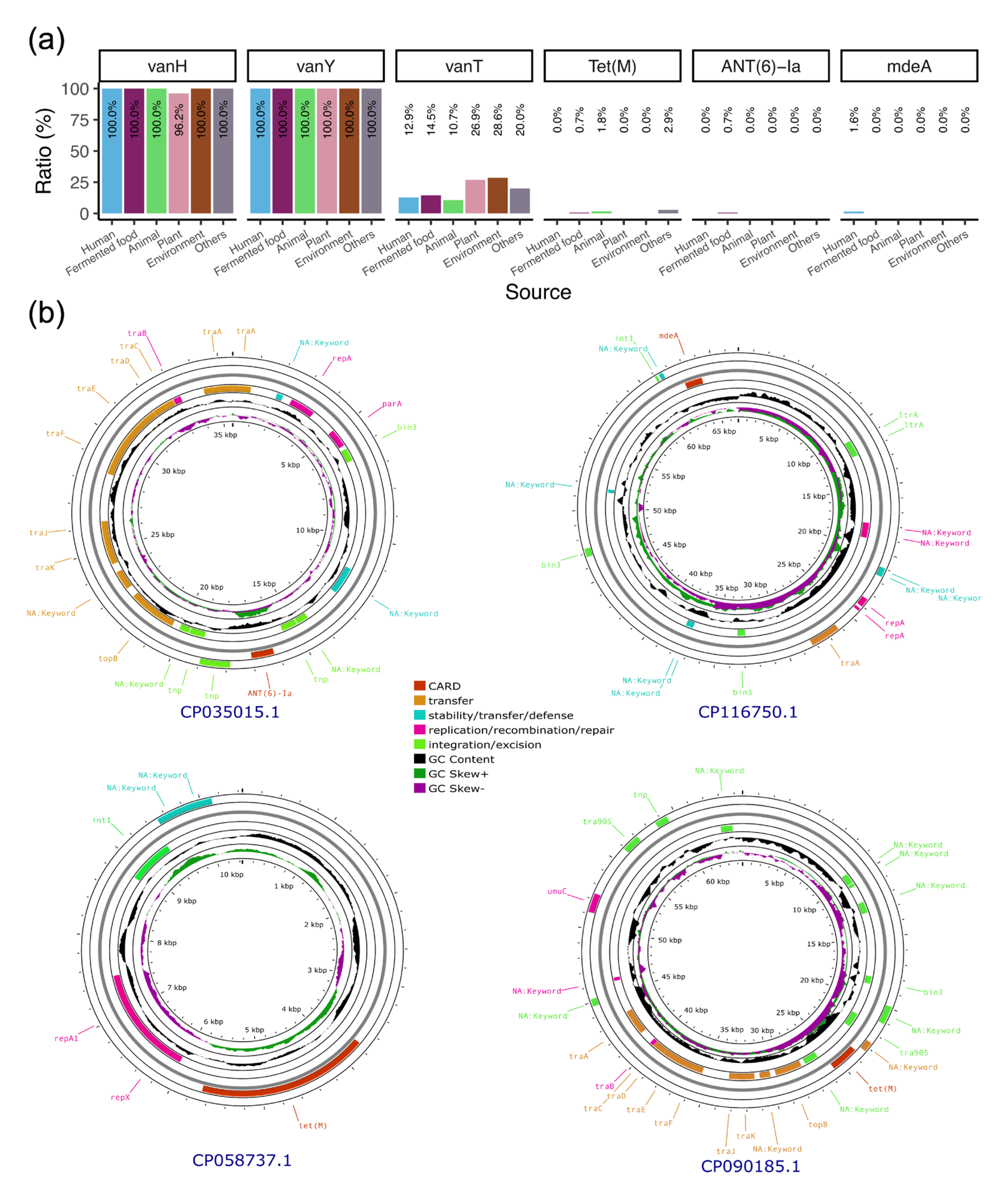
3.7. Plasmids Mediate the Transmission of Antibiotic Resistance Genes Within the Lactobacillaceae Family
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMP | Antimicrobial Peptide |
| ANI | Average Nucleotide Identity |
| ARG | Antibiotic Resistance Gene |
| CARD | Comprehensive Antibiotic Resistance Database |
| CAZyme | Carbohydrate-Active Enzyme |
| GH | Glycoside Hydrolase |
| IMP | Inosine Monophosphate |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MGE | Mobile Genetic Element |
| MIC | Minimum Inhibitory Concentration |
| NCBI | National Center for Biotechnology Information |
References
- Siezen, R.J.; van Hylckama Vlieg, J.E.T. Genomic Diversity and Versatility of Lactobacillus plantarum, a Natural Metabolic Engineer. Microb. Cell Fact. 2011, 10 (Suppl. S1), S3. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.B.; Baiseitova, A.; Zahoor, M.; Ahmad, I.; Ikram, M.; Bakhsh, A.; Shah, M.A.; Ali, I.; Idress, M.; Ullah, R.; et al. Probiotic Significance of Lactobacillus Strains: A Comprehensive Review on Health Impacts, Research Gaps, and Future Prospects. Gut Microbes 2024, 16, 2431643. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wittouck, S.; Salvetti, E.; Franz, C.M.A.P.; Harris, H.M.B.; Mattarelli, P.; O’Toole, P.W.; Pot, B.; Vandamme, P.; Walter, J.; et al. A Taxonomic Note on the Genus Lactobacillus: Description of 23 Novel Genera, Emended Description of the Genus Lactobacillus Beijerinck 1901, and Union of Lactobacillaceae and Leuconostocaceae. Int. J. Syst. Evol. Microbiol. 2020, 70, 2782–2858. [Google Scholar] [CrossRef]
- Chen, K.; Luan, X.; Liu, Q.; Wang, J.; Chang, X.; Snijders, A.M.; Mao, J.-H.; Secombe, J.; Dan, Z.; Chen, J.-H.; et al. Drosophila Histone Demethylase KDM5 Regulates Social Behavior through Immune Control and Gut Microbiota Maintenance. Cell Host Microbe 2019, 25, 537–552.e8. [Google Scholar] [CrossRef]
- Cui, Y.; Wang, M.; Zheng, Y.; Miao, K.; Qu, X. The Carbohydrate Metabolism of Lactiplantibacillus plantarum. Int. J. Mol. Sci. 2021, 22, 13452. [Google Scholar] [CrossRef]
- Tegopoulos, K.; Stergiou, O.S.; Kiousi, D.E.; Tsifintaris, M.; Koletsou, E.; Papageorgiou, A.C.; Argyri, A.A.; Chorianopoulos, N.; Galanis, A.; Kolovos, P. Genomic and Phylogenetic Analysis of Lactiplantibacillus plantarum L125, and Evaluation of Its Anti-Proliferative and Cytotoxic Activity in Cancer Cells. Biomedicines 2021, 9, 1718. [Google Scholar] [CrossRef]
- Huidrom, S.; Ngashangva, N.; Khumlianlal, J.; Sharma, K.C.; Mukherjee, P.K.; Devi, S.I. Genomic Insights from Lactiplantibacillus plantarum BRD3A Isolated from Atingba, a Traditional Fermented Rice-Based Beverage and Analysis of Its Potential for Probiotic and Antimicrobial Activity against Methicillin-Resistant Staphylococcus Aureus. Front. Microbiol. 2024, 15, 1357818. [Google Scholar] [CrossRef]
- Tóth, A.G.; Judge, M.F.; Nagy, S.Á.; Papp, M.; Solymosi, N. A Survey on Antimicrobial Resistance Genes of Frequently Used Probiotic Bacteria, 1901 to 2022. Eurosurveillance 2023, 28, 2200272. [Google Scholar] [CrossRef]
- Garcia-Gonzalez, N.; Bottacini, F.; van Sinderen, D.; Gahan, C.G.M.; Corsetti, A. Comparative Genomics of Lactiplantibacillus plantarum: Insights Into Probiotic Markers in Strains Isolated From the Human Gastrointestinal Tract and Fermented Foods. Front. Microbiol. 2022, 13, 854266. [Google Scholar] [CrossRef]
- Evanovich, E.; de Souza Mendonça Mattos, P.J.; Guerreiro, J.F. Comparative Genomic Analysis of Lactobacillus plantarum: An Overview. Int. J. Genom. 2019, 2019, 4973214. [Google Scholar] [CrossRef]
- Wu, M.; Tarrah, A.; Ghion, G.; Pakroo, S.; Giacomini, A.; Corich, V. A Critical Issue on Microbiological Cut-off Value of Ampicillin Resistance in Lactiplantibacillus plantarum. J. Appl. Microbiol. 2023, 134, lxad050. [Google Scholar] [CrossRef]
- Martino, M.E.; Bayjanov, J.R.; Caffrey, B.E.; Wels, M.; Joncour, P.; Hughes, S.; Gillet, B.; Kleerebezem, M.; van Hijum, S.A.F.T.; Leulier, F. Nomadic Lifestyle of Lactobacillus plantarum Revealed by Comparative Genomics of 54 Strains Isolated from Different Habitats. Environ. Microbiol. 2016, 18, 4974–4989. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and Taxonomy in Diagnostics for Food Security: Soft-Rotting Enterobacterial Plant Pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Tonkin-Hill, G.; MacAlasdair, N.; Ruis, C.; Weimann, A.; Horesh, G.; Lees, J.A.; Gladstone, R.A.; Lo, S.; Beaudoin, C.; Floto, R.A.; et al. Producing Polished Prokaryotic Pangenomes with the Panaroo Pipeline. Genome Biol. 2020, 21, 180. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. EggNOG 5.0: A Hierarchical, Functionally and Phylogenetically Annotated Orthology Resource Based on 5090 Organisms and 2502 Viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R.; Teeling, E. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Tonkin-Hill, G.; Lees, J.A.; Bentley, S.D.; Frost, S.D.W.; Corander, J. Fast Hierarchical Bayesian Analysis of Population Structure. Nucleic Acids Res. 2019, 47, 5539–5549. [Google Scholar] [CrossRef]
- Zhou, Z.; Tran, P.Q.; Breister, A.M.; Liu, Y.; Kieft, K.; Cowley, E.S.; Karaoz, U.; Anantharaman, K. METABOLIC: High-Throughput Profiling of Microbial Genomes for Functional Traits, Metabolism, Biogeochemistry, and Community-Scale Functional Networks. Microbiome 2022, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Liu, B.; Zheng, D.; Chen, L.; Yang, J. VFDB 2025: An Integrated Resource for Exploring Anti-Virulence Compounds. Nucleic Acids Res. 2025, 53, D871–D877. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Huynh, W.; Chalil, R.; Smith, K.W.; Raphenya, A.R.; Wlodarski, M.A.; Edalatmand, A.; Petkau, A.; Syed, S.A.; Tsang, K.K.; et al. CARD 2023: Expanded Curation, Support for Machine Learning, and Resistome Prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023, 51, D690–D699. [Google Scholar] [CrossRef]
- Camargo, A.P.; Roux, S.; Schulz, F.; Babinski, M.; Xu, Y.; Hu, B.; Chain, P.S.G.; Nayfach, S.; Kyrpides, N.C. Identification of Mobile Genetic Elements with geNomad. Nat. Biotechnol. 2023, 42, 1303–1312. [Google Scholar] [CrossRef]
- Zheng, K.; Sun, J.; Liang, Y.; Kong, L.; Paez-Espino, D.; Mcminn, A.; Wang, M. VITAP: A High Precision Tool for DNA and RNA Viral Classification Based on Meta-Omic Data. Nat. Commun. 2025, 16, 2226. [Google Scholar] [CrossRef]
- Robertson, J.; Bessonov, K.; Schonfeld, J.; Nash, J.H.E. Universal Whole-Sequence-Based Plasmid Typing and Its Utility to Prediction of Host Range and Epidemiological Surveillance. Microb. Genom. 2020, 6, mgen000435. [Google Scholar] [CrossRef]
- Santos-Júnior, C.D.; Pan, S.; Zhao, X.-M.; Coelho, L.P. Macrel: Antimicrobial Peptide Screening in Genomes and Metagenomes. PeerJ 2020, 8, e10555. [Google Scholar] [CrossRef]
- Mondal, R.K.; Sen, D.; Arya, A.; Samanta, S.K. Developing Anti-Microbial Peptide Database Version 1 to Provide Comprehensive and Exhaustive Resource of Manually Curated AMPs. Sci. Rep. 2023, 13, 17843. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The Antimicrobial Peptide Database as a Tool for Research and Education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef]
- Pirtskhalava, M.; Amstrong, A.A.; Grigolava, M.; Chubinidze, M.; Alimbarashvili, E.; Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M. DBAASP v3: Database of Antimicrobial/Cytotoxic Activity and Structure of Peptides as a Resource for Development of New Therapeutics. Nucleic Acids Res. 2021, 49, D288–D297. [Google Scholar] [CrossRef]
- Shi, G.; Kang, X.; Dong, F.; Liu, Y.; Zhu, N.; Hu, Y.; Xu, H.; Lao, X.; Zheng, H. DRAMP 3.0: An Enhanced Comprehensive Data Repository of Antimicrobial Peptides. Nucleic Acids Res. 2022, 50, D488–D496. [Google Scholar] [CrossRef]
- Ye, G.; Wu, H.; Huang, J.; Wang, W.; Ge, K.; Li, G.; Zhong, J.; Huang, Q. LAMP2: A Major Update of the Database Linking Antimicrobial Peptides. Database 2020, 2020, baaa061. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Torres, M.D.T.; Peng, J.; de la Fuente-Nunez, C. Deep-Learning-Enabled Antibiotic Discovery through Molecular de-Extinction. Nat. Biomed. Eng. 2024, 8, 854–871. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v6: Recent Updates to the Phylogenetic Tree Display and Annotation Tool. Nucleic Acids Res. 2024, 52, W78–W82. [Google Scholar] [CrossRef]
- Grant, J.R.; Enns, E.; Marinier, E.; Mandal, A.; Herman, E.K.; Chen, C.-Y.; Graham, M.; Van Domselaar, G.; Stothard, P. Proksee: In-Depth Characterization and Visualization of Bacterial Genomes. Nucleic Acids Res. 2023, 51, W484–W492. [Google Scholar] [CrossRef]
- Kolde, R.; Kolde, M.R. Maintainer Raivo Package ‘pheatmap’. Bioconductor 2015, 1, 790. [Google Scholar]
- Brunson, J.C. Ggalluvial: Alluvial Plots in “Ggplot2”. ggalluvial 2020, 5, 2017. [Google Scholar]
- Zhang, L.; Kulyar, M.F.; Niu, T.; Yang, S.; Chen, W. Comparative Genomics of Limosilactobacillus Reuteri YLR001 Reveals Genetic Diversity and Probiotic Properties. Microorganisms 2024, 12, 1636. [Google Scholar] [CrossRef]
- You, L.; Lv, R.; Jin, H.; Ma, T.; Zhao, Z.; Kwok, L.-Y.; Sun, Z. A Large-Scale Comparative Genomics Study Reveals Niche-Driven and within-Sample Intra-Species Functional Diversification in Lacticaseibacillus rhamnosus. Food Res. Int. 2023, 173, 113446. [Google Scholar] [CrossRef] [PubMed]
- Myrbråten, I.S.; Wiull, K.; Salehian, Z.; Håvarstein, L.S.; Straume, D.; Mathiesen, G.; Kjos, M. CRISPR Interference for Rapid Knockdown of Essential Cell Cycle Genes in Lactobacillus plantarum. mSphere 2019, 4, e00007–e00019. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Liu, W.; Piao, M.; Zhu, H. A Review of the Relationship between the Gut Microbiota and Amino Acid Metabolism. Amino Acids 2017, 49, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Man, L.-L.; Xiang, D.-J. Effect of LuxS/AI-2-Mediated Quorum Sensing System on Bacteriocin Production of Lactobacillus plantarum NMD-17. Folia Microbiol. 2023, 68, 855–866. [Google Scholar] [CrossRef]
- Hu, Q.; Qi, Y.; Liu, C.; Chen, Q.; Mai, X.; Zhu, Z.; Ma, B. Effects of Lactobacillus plantarum Cofermentation on the Flavor and Taste Characteristics of Mango Vinegar. J. Food Meas. Charact. 2024, 18, 3744–3756. [Google Scholar] [CrossRef]
- Mann, E.R.; Lam, Y.K.; Uhlig, H.H. Short-Chain Fatty Acids: Linking Diet, the Microbiome and Immunity. Nat. Rev. Immunol. 2024, 24, 577–595. [Google Scholar] [CrossRef]
- Gonçalves, C.; Gonçalves, P. Multilayered Horizontal Operon Transfers from Bacteria Reconstruct a Thiamine Salvage Pathway in Yeasts. Proc. Natl. Acad. Sci. USA 2019, 116, 22219–22228. [Google Scholar] [CrossRef]
- Iarusso, I.; Mahony, J.; Pannella, G.; Lombardi, S.J.; Gagliardi, R.; Coppola, F.; Pellegrini, M.; Succi, M.; Tremonte, P. Diversity of Lactiplantibacillus plantarum in Wild Fermented Food Niches. Foods 2025, 14, 1765. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Cut-Off | Number of Genes |
|---|---|---|
| Core genes | 99% ≤ strains ≤ 100% | 2194 |
| Soft-core genes | 95% ≤ strains < 99% | 209 |
| Shell genes | 15% ≤ strains < 95% | 1164 |
| Cloud genes | 0% < strains < 15% | 8849 |
| Total genes | 0% < strains ≤ 100% | 12,416 |
| Accession | Strain Host | ARG | Replicon Type | Predicted Mobility | Predicted Host Range |
|---|---|---|---|---|---|
| CP035015 | L. plantarum 12_3 | ANT(6)-Ia | rep_cluster_707 | Conjugative | Lactobacillaceae |
| CP058737 | L. plantarum A8 | Tet(M) | rep_cluster_2119 | Non-mobilizable | Lactobacillaceae |
| CP090185 | L. plantarum ST | Tet(M) | rep_cluster_167, rep_cluster_707 | Conjugative | Lactobacillaceae |
| CP116750 | L. plantarum MWLp-12 | mdeA | rep_cluster_1328 | Mobilizable | Lactobacillales |
| CP140093 | L. plantarum J50 | Tet(M) | rep_cluster_2119 | Non-mobilizable | Lactobacillaceae |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Bi, C. Comparative Genomic Analysis of Lactiplantibacillus plantarum: Insights into Its Genetic Diversity, Metabolic Function, and Antibiotic Resistance. Genes 2025, 16, 869. https://doi.org/10.3390/genes16080869
Li R, Bi C. Comparative Genomic Analysis of Lactiplantibacillus plantarum: Insights into Its Genetic Diversity, Metabolic Function, and Antibiotic Resistance. Genes. 2025; 16(8):869. https://doi.org/10.3390/genes16080869
Chicago/Turabian StyleLi, Ruiqi, and Chongpeng Bi. 2025. "Comparative Genomic Analysis of Lactiplantibacillus plantarum: Insights into Its Genetic Diversity, Metabolic Function, and Antibiotic Resistance" Genes 16, no. 8: 869. https://doi.org/10.3390/genes16080869
APA StyleLi, R., & Bi, C. (2025). Comparative Genomic Analysis of Lactiplantibacillus plantarum: Insights into Its Genetic Diversity, Metabolic Function, and Antibiotic Resistance. Genes, 16(8), 869. https://doi.org/10.3390/genes16080869