
Two Years of Growth Hormone Therapy in a Child with Severe Short Stature Due to Overlap Syndrome with a Novel SETD5 Gene Mutation: Case Report and Review of the Literature

 ,
,  ,
,  , , , , and
, , , , and 
Abstract
1. Introduction
2. Case Report
3. Discussion
3.1. SETD5 Gene Mutation and Genetic Overlap Syndrome
3.2. Focus on Growth Hormone Therapy and Growth Rate in CdL and KBG Syndromes
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cohen, L.E.; Alan, D.R. Children With Idiopathic Short Stature: An Expanding Role for Genetic Investigation in Their Medical Evaluation. Endocr. Pract. 2024, 30, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Zhou, E.; Hauser, B.R.; Jee, Y.H. Genetic evaluation in children with short stature. Curr. Opin. Pediatr. 2021, 33, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Danowitz, M.; Grimberg, A. Clinical Indications for Growth Hormone Therapy. Adv. Pediatr. 2022, 69, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Ranke, M.B.; Wit, J.M. Growth hormone—Past, present and future. Nat. Rev. Endocrinol. 2018, 14, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Agenzia Italiana del Farmaco (AIFA). *Nota 39: Prescrizione di Ormone Della Crescita (Somatotropina—GH)*. Modifica Della Determina n. 431/2021 (GU Serie Generale 74, 28 Marzo 2023). 2023. Available online: https://www.gazzettaufficiale.it/atto/serie_generale/caricaArticolo?art.versione=1&art.idGruppo=0&art.flagTipoArticolo=1&art.codiceRedazionale=23A01960&art.idArticolo=1&art.idSottoArticolo=1&art.idSottoArticolo1=10&art.dataPubblicazioneGazzetta=2023-03-28&art.progressivo=0 (accessed on 16 July 2025).
- Takeda, A.; Cooper, K.; Bird, A.; Baxter, L.; Frampton, G.K.; Gospodarevskaya, E.; Welch, K.; Bryant, J. Recombinant human growth hormone for the treatment of growth disorders in children: A systematic review and economic evaluation. Health Technol. Assess. 2010, 14, i–iv. [Google Scholar] [CrossRef] [PubMed]
- Kucharska, A.; Witkowska-Sędek, E.; Erazmus, M.; Artemniak-Wojtowicz, D.; Krajewska, M.; Pyrżak, B. The Effects of Growth Hormone Treatment Beyond Growth Promotion in Patients with Genetic Syndromes: A Systematic Review of the Literature. Int. J. Mol. Sci. 2024, 25, 10169. [Google Scholar] [CrossRef] [PubMed]
- Powis, Z.; Farwell Hagman, K.D.; Mroske, C.; McWalter, K.; Cohen, J.S.; Colombo, R.; Serretti, A.; Fatemi, A.; David, K.L.; Reynolds, J.; et al. Expansion and further delineation of the SETD5 phenotype leading to global developmental delay, variable dysmorphic features, and reduced penetrance. Clin. Genet. 2018, 93, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Pascolini, G.; Gnazzo, M.; Novelli, A.; Grammatico, P. Clinical refinement of the SETD5-associated phenotype in a child displaying novel features and KBG syndrome-like appearance. Am. J. Med. Genet. A 2022, 188, 1623–1625. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, M.; Kant, S.G.; Wit, J.M.; Willem Redeker, E.J.; Eduard Santen, G.W.; Henriëtta Verkerk, A.J.M.; Uitterlinden, A.G.; Losekoot, M.; Oostdijk, W. Successful Growth Hormone Therapy in Cornelia de Lange Syndrome. J. Clin. Res. Pediatr. Endocrinol. 2017, 9, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Reynaert, N.; Ockeloen, C.W.; Sävendahl, L.; Beckers, D.; Devriendt, K.; Kleefstra, T.; Carels, C.E.; Grigelioniene, G.; Nordgren, A.; Francois, I.; et al. Short Stature in KBG Syndrome: First Responses to Growth Hormone Treatment. Horm. Res. Paediatr. 2015, 83, 361–364. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; He, D.; Li, Y.; Zhang, Y.; Shao, Q.; Zhang, M.; Ban, B. A heterozygous point mutation of the ANKRD11 (c.2579C>T) in a Chinese patient with idiopathic short stature. Mol. Genet. Genomic Med. 2019, 7, e988. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.Y.; Ge, L.; Hu, W.W.; Li, X.L.; Hu, Y.Y. Growth hormone therapy for children with KBG syndrome: A case report and review of literature. World J. Clin. Cases. 2020, 8, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Hörenz, C.; Vogel, M.; Wirkner, K.; Ceglarek, U.; Thiery, J.; Pfäffle, R.; Kiess, W.; Kratzsch, J. BMI and Contraceptives Affect New Age-, Sex-, and Puberty-adjusted IGF-I and IGFBP-3 Reference Ranges Across Life Span. J. Clin. Endocrinol. Metab. 2022, 107, e2991–e3002. [Google Scholar] [CrossRef] [PubMed]
- Cacciari, E.; Milani, S.; Balsamo, A.; Dammacco, F.; De Luca, F.; Chiarelli, F.; Pasquino, A.M.; Tonini, G.; Vanelli, M. Italian cross-sectional growth charts for height, weight and BMI (6–20 y). Eur. J. Clin. Nutr. 2002, 56, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J.M.; Davies, P.S. Clinical longitudinal standards for height and height velocity for North American children. J. Pediatr. 1985, 107, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Deliu, E.; Arecco, N.; Morandell, J.; Dotter, C.P.; Contreras, X.; Girardot, C.; Käsper, E.L.; Kozlova, A.; Kishi, K.; Chiaradia, I.; et al. Haploinsufficiency of the intellectual disability gene SETD5 disturbs developmental gene expression and cognition. Nat. Neurosci. 2018, 21, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Hattori, S.; Hosoi, T.; Nakayama, K. Neurobehavioral characteristics of mice with SETD5mutations as models of IDD23 and KBG syndromes. Front. Genet. 2023, 13, 1022339. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, I.R.; Cruz, A.C.P.; Ferrasa, A.; Phan, D.; Herai, R.H.; Muotri, A.R. Genetic variations on SETD5 underlying autistic conditions. Dev. Neurobiol. 2018, 78, 500–518. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, H.; Wang, J.; Lin, J.; Huang, X.; Zeng, Y.; Chen, R. A study on genotypes and phenotypes of short stature caused by epigenetic modification gene variants. Eur. J. Pediatr. 2024, 183, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Grozeva, D.; Carss, K.; Spasic-Boskovic, O.; Parker, M.J.; Archer, H.; Firth, H.V.; Park, S.M.; Canham, N.; Holder, S.E.; Wilson, M.; et al. De novo loss-of-function mutations in SETD5, encoding a methyltransferase in a 3p25 microdeletion syndrome critical region, cause intellectual disability. Am. J. Hum. Genet. 2014, 94, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wang, T.; Fu, Z.; Li, T.; Zhang, X.; Zhao, J.; Yang, G. Case Report: A Case Report and Literature Review of 3p Deletion Syndrome. Front. Pediatr. 2021, 9, 618059. [Google Scholar] [CrossRef] [PubMed]
- Kuechler, A.; Zink, A.M.; Wieland, T.; Lüdecke, H.J.; Cremer, K.; Salviati, L.; Magini, P.; Najafi, K.; Zweier, C.; Czeschik, J.C.; et al. Loss-of-function variants of SETD5 cause intellectual disability and the core phenotype of microdeletion 3p25.3 syndrome. Eur. J. Hum. Genet. 2015, 23, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Stur, E.; Soares, L.A.; Louro, I.D. SETD5 gene variant associated with mild intellectual disability—A case report. Genet. Mol. Res. 2017, 16, 3737–3740. [Google Scholar] [CrossRef] [PubMed]
- Peltekova, I.T.; Macdonald, A.; Armour, C.M. Microdeletion on 3p25 in a patient with features of 3p deletion syndrome. Am. J. Med. Genet. A 2012, 158A, 2583–2586. [Google Scholar] [CrossRef] [PubMed]
- Kline, A.D.; Moss, J.F.; Selicorni, A.; Bisgaard, A.M.; Deardorff, M.A.; Gillett, P.M.; Ishman, S.L.; Kerr, L.; Levin, A.V.; Mulder, P.A.; et al. Diagnosis and management of Cornelia de Lange syndrome: First international consensus statement. Nat. Rev. Genet. 2018, 19, 649–666. [Google Scholar] [CrossRef] [PubMed]
- Parenti, I.; Kaiser, F.J. Cornelia de Lange Syndrome as Paradigm of Chromatinopathies. Front. Neurosci. 2021, 15, 774950. [Google Scholar] [CrossRef] [PubMed]
- Shangguan, H.; Chen, R. Phenotypes of Cornelia de Lange syndrome caused by non-cohesion genes: Novel variants and literature review. Front. Pediatr. 2022, 10, 940294. [Google Scholar] [CrossRef] [PubMed]
- Parenti, I.; Teresa-Rodrigo, M.E.; Pozojevic, J.; Ruiz Gil, S.; Bader, I.; Braunholz, D.; Bramswig, N.C.; Gervasini, C.; Larizza, L.; Pfeiffer, L.; et al. Mutations in chromatin regulators functionally link Cornelia de Lange syndrome and clinically overlapping phenotypes. Hum. Genet. 2017, 136, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Morel Swols, D.; Tekin, M. KBG Syndrome; 2018 Mar. GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2018. Available online: https://www.ncbi.nlm.nih.gov/books/NBK487886/ (accessed on 29 June 2025).
- Sirmaci, A.; Spiliopoulos, M.; Brancati, F.; Powell, E.; Duman, D.; Abrams, A.; Bademci, G.; Agolini, E.; Guo, S.; Konuk, B.; et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am. J. Hum. Genet. 2011, 89, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Crippa, M.; Bestetti, I.; Maitz, S.; Weiss, K.; Spano, A.; Masciadri, M.; Smithson, S.; Larizza, L.; Low, K.; Cohen, L.; et al. SETD5 Gene Haploinsufficiency in Three Patients With Suspected KBG Syndrome. Front. Neurol. 2020, 11, 631. [Google Scholar] [CrossRef] [PubMed]
- Maghnie, M.; Ranke, M.B.; Geffner, M.E.; Vlachopapadopoulou, E.; Ibáñez, L.; Carlsson, M.; Cutfield, W.; Rooman, R.; Gomez, R.; Wajnrajch, M.P.; et al. Safety and Efficacy of Pediatric Growth Hormone Therapy: Results From the Full KIGS Cohort. J. Clin. Endocrinol. Metab. 2022, 107, 3287–3301. [Google Scholar] [CrossRef] [PubMed]
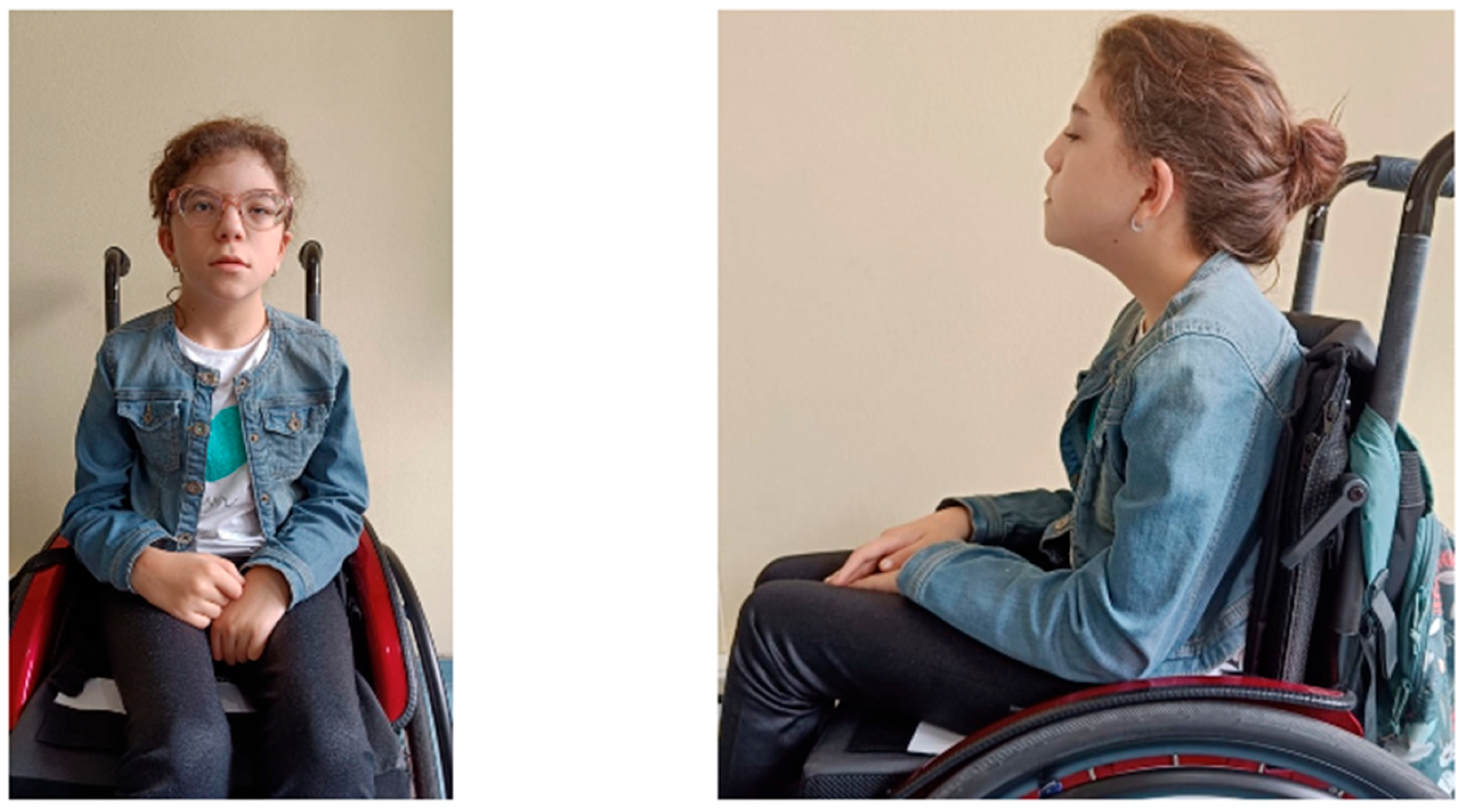

{kind=link}
{kind=link}
| KBG syndrome (KBGS) is characterized by developmental delay/learning difficulties, speech delay, significant behavioral issue and at least two major criteria or one major and two minor criteria | Classic Cornelia de Lange syndrome (CdLS) is defined with a clinical score ≥ 11 points, of which at least 3 are cardinal features. Non-classic CdLS presents a clinical score of 9-10 points, of which at least 2 are cardinal features. Molecular testing for CdLS is indicated with a clinical score of 4-8 points. |
| Major Criteria: Macrodontia (large teeth) Recurrent otitis media and/or hearing loss. Post-natal short stature (height <10th percentile) First-degree relative with KBG syndrome | Cardinal features (2 points each of present): Synophrys and/or thick eyebrows Short nose, concave nasale ridge, and/or upturned nasal tip Hand oligodactyly and/or adactyly Long and/or smooth philtrum Thin upper lip vermilion and/or downturned corners of mouth Congenital diaphragmatic hernia |
| Minor Criteria: Brachydactyly (short fingers) or hand anomaly Cryptorchidism (undescended testes) Seizures Feeding problems Palate abnormality Large anterior fontanelle and/or delayed closure Autism diagnosis | Suggestive features (1 point each if present): Global developmental delay and/or intellectual disability Short fifth finger Prenatal growth retardation Small hands and/or feet Postnatal growth retardation Hirsutism |
| Clinical Features of Our Patient | MDR23 | CdLS | KBGS |
|---|---|---|---|
| Intellectual disability |  |  |  |
| Delayed speech development |  |  | |
| Hypotonia |  | ||
| Short stature and postnatal growth retardation |  |  |  |
| Dysmorphic feature |  |  |  |
| Macrodontia |  | ||
| Synophrys |  | ||
| Short nose and concave nasal ridge |  | ||
| Long philtrum |  |
| References | Case/Syndrome/and Additional Diseases | Genetic Variant | Age of Start and End of rHGh Therapy | Dose of rhGH | Height SDS at the Beginning of Treatment | Height SDS at the End of Treatment | Comments |
|---|---|---|---|---|---|---|---|
| De Graaf et al. [10] | Female patient born small for gestational age and with CdL syndrome | c.771+1G>A (chr5:36971139) in NIPBL gene | From 4.3 years to 8 years | 0.86 mg/m2/day | −3.5 SDS | −1.4 SDS | rhGH therapy was indicated for small for gestational age. Increase in height SDS of 1.8 SDS following treatment with r-hGH |
| Reynaert et al. [11] | Male with KBG syndrome | c.3836del (p.Ser1279fs) in ANKRD11 gene | From 10.5 years. Last evaluation when he was 13.8 years | 35 μg/kg/day | −3.1 SDS | −1.6 SDS | Triptorelin treatment due to medical necessity was started at 12.5 years (height −1.7 SDS) because of the fast progression of puberty. At age 13.8 years triptorelin was stopped. |
| Male with KBG Syndrome | c.1903_1907del (p.Lys635Glnf*26) in ANKRD1 gene | From 7.4 years. Last evaluation when he was 12.4 years | 30 μg/kg/day | −2.8 SDS | −0.7 SDS | The patient’s height increased by 1.0 SD during the first year on treatment and by another 1.1 SD in the subsequent 4 years. | |
| Xiu-Ying Ge et al. [12] | Girl with Growth hormone deficiencies and KBG syndrome | c.2635 dupG, (p.Glu879fs) in ANKRD11 gene | From 5.6 years to 7.6 years | 50 μg/kg/day | −1.95 SDS | −0.70 SDS | rhGH therapy was indicated for Growth hormone deficiencies. |
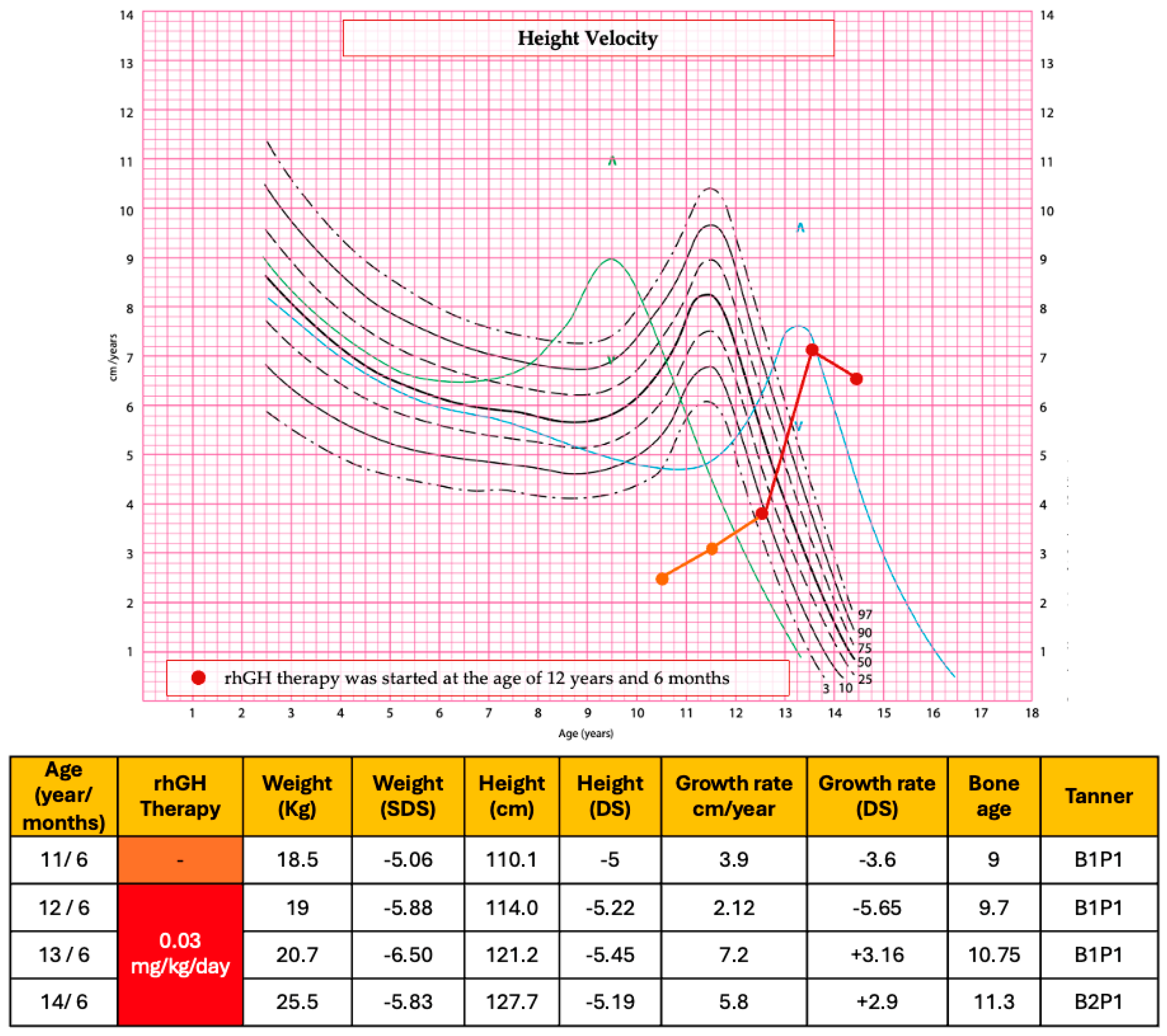
| Our patient | Girl with overlap syndrome (MDR23, CdL and KBG syndrome) | c.890_891delTT (p.Leu297fs*15) in SETD5 gene | From 12.6 years to 14.6 years | 30 μg/kg/day | −5.22 SDS | −5.19 SDS * | After the first year of therapy, the growth rate had been 7.2 cm (+3.16 SDS). After 2 years of treatment, at the age of 14 years and 6 months, the growth rate had been 5.8 cm/year (+2.9 SDS). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luppino, G.; Wasniewska, M.; Pepe, G.; Morabito, L.A.; Briuglia, S.; Moschella, A.; Franchina, F.; Lugarà, C.; Aversa, T.; Corica, D. Two Years of Growth Hormone Therapy in a Child with Severe Short Stature Due to Overlap Syndrome with a Novel SETD5 Gene Mutation: Case Report and Review of the Literature. Genes 2025, 16, 859. https://doi.org/10.3390/genes16080859
Luppino G, Wasniewska M, Pepe G, Morabito LA, Briuglia S, Moschella A, Franchina F, Lugarà C, Aversa T, Corica D. Two Years of Growth Hormone Therapy in a Child with Severe Short Stature Due to Overlap Syndrome with a Novel SETD5 Gene Mutation: Case Report and Review of the Literature. Genes. 2025; 16(8):859. https://doi.org/10.3390/genes16080859
Chicago/Turabian StyleLuppino, Giovanni, Malgorzata Wasniewska, Giorgia Pepe, Letteria Anna Morabito, Silvana Briuglia, Antonino Moschella, Francesca Franchina, Cecilia Lugarà, Tommaso Aversa, and Domenico Corica. 2025. "Two Years of Growth Hormone Therapy in a Child with Severe Short Stature Due to Overlap Syndrome with a Novel SETD5 Gene Mutation: Case Report and Review of the Literature" Genes 16, no. 8: 859. https://doi.org/10.3390/genes16080859
APA StyleLuppino, G., Wasniewska, M., Pepe, G., Morabito, L. A., Briuglia, S., Moschella, A., Franchina, F., Lugarà, C., Aversa, T., & Corica, D. (2025). Two Years of Growth Hormone Therapy in a Child with Severe Short Stature Due to Overlap Syndrome with a Novel SETD5 Gene Mutation: Case Report and Review of the Literature. Genes, 16(8), 859. https://doi.org/10.3390/genes16080859