
Patchy Phylogenetic Distribution and Poor Translational Adaptation of a Nested ORF in the Mammalian Mitochondrial cytb Gene


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequence Data Acquisition
2.2. Screening for Nested Open Reading Frames
2.3. Homology Analysis of ncytb-Encoded Proteins
2.4. Multiple Sequence Alignment of ncytb-Encoded Proteins
2.5. Phylogenetic Tree of cytb-Encoded Proteins
2.6. Analysis of Translational Context
2.7. Prediction of ncytb-Encoded Protein Properties
- MitoFates: https://mitf.cbrc.pj.aist.go.jp/MitoFates/cgi-bin/top.cgi, accessed on 4 December 2024, used with default parameters for mitochondrial presequence prediction [15];
- SignalP-6.0: https://services.healthtech.dtu.dk/services/SignalP-6.0/, accessed on 28 December 2024, used with the “Mode: slow” model for signal peptide prediction [16];
- DeepLoc-2.1: https://services.healthtech.dtu.dk/services/DeepLoc-2.1/, accessed on 28 December 2024, used with the “High-quality (Slow)” model for subcellular localization prediction [13];
- DeepTMHMM 1.0: https://services.healthtech.dtu.dk/services/DeepTMHMM-1.0/, accessed on 29 December 2024, used with default parameters for transmembrane region prediction [17];
- NCBI Conserved Domain Database (CDD): https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi, accessed on 29 December 2024, searched with default parameters for functional domain identification [18];
- IPC 2.0: https://ipc2.mimuw.edu.pl/, accessed on 4 January 2025, used with default parameters for isoelectric point (pI) prediction [19].
2.8. Codon Adaptation Index (CAI)
2.9. Relative Codon Deoptimization Index (RCDI)
2.10. Cosine Similarity Analysis
3. Results
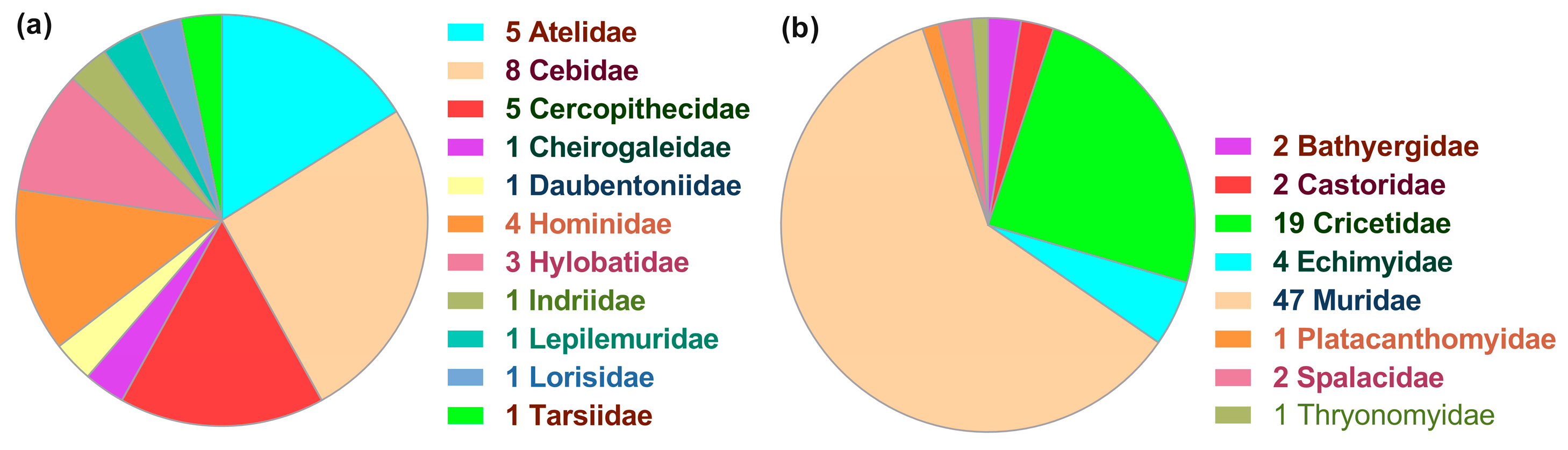
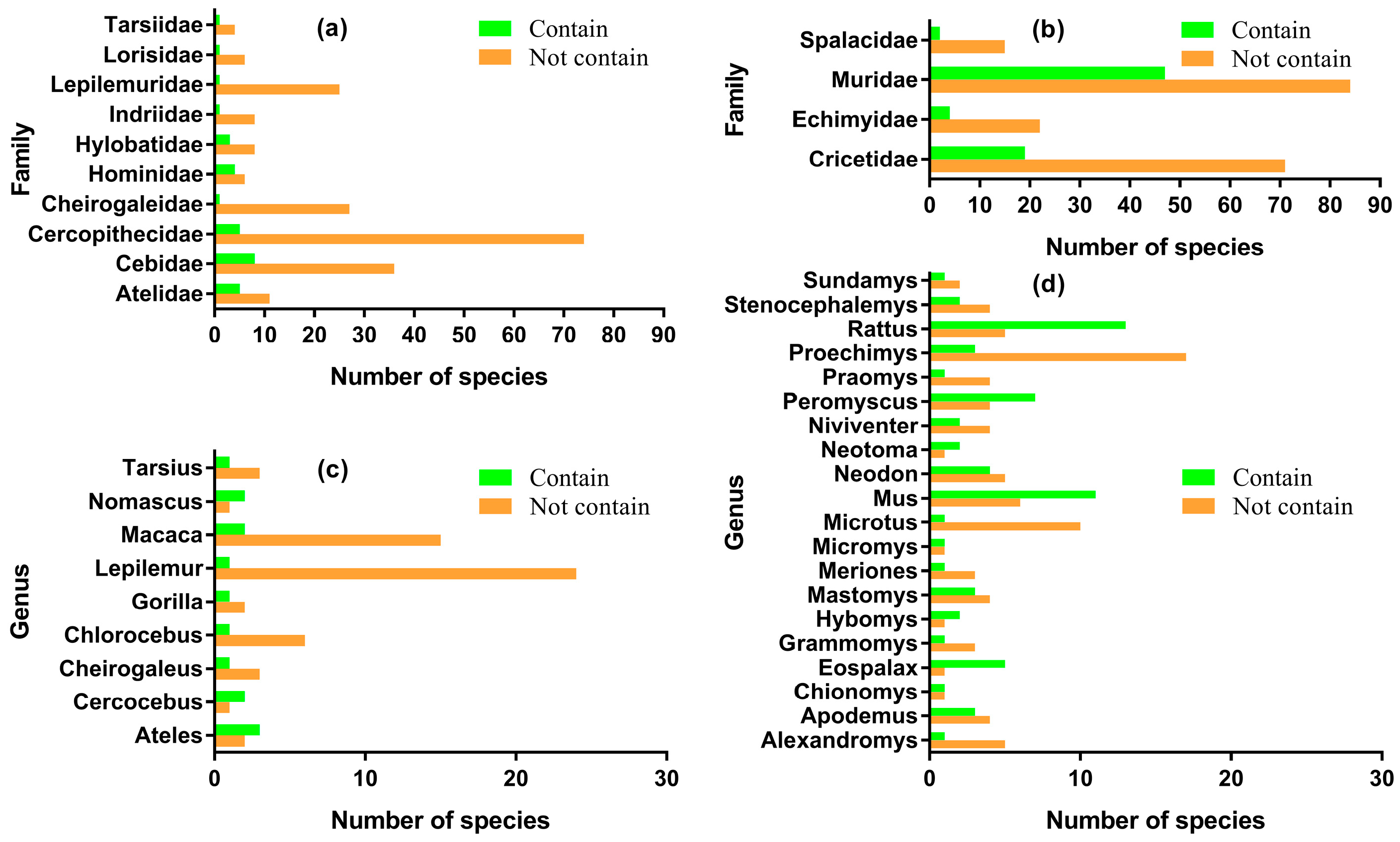
3.1. Patchy Phylogenetic Distribution of the Nested ncytb Gene
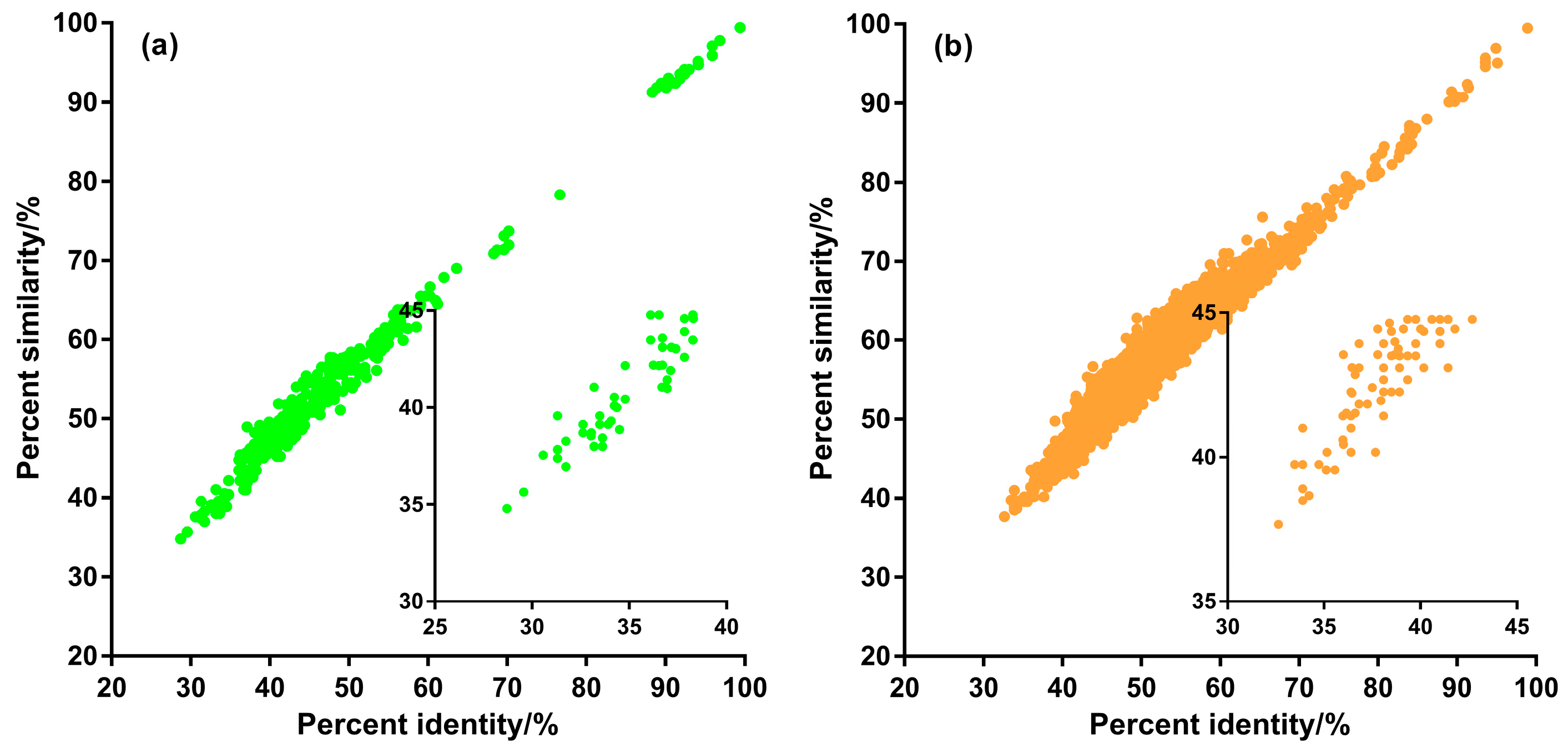
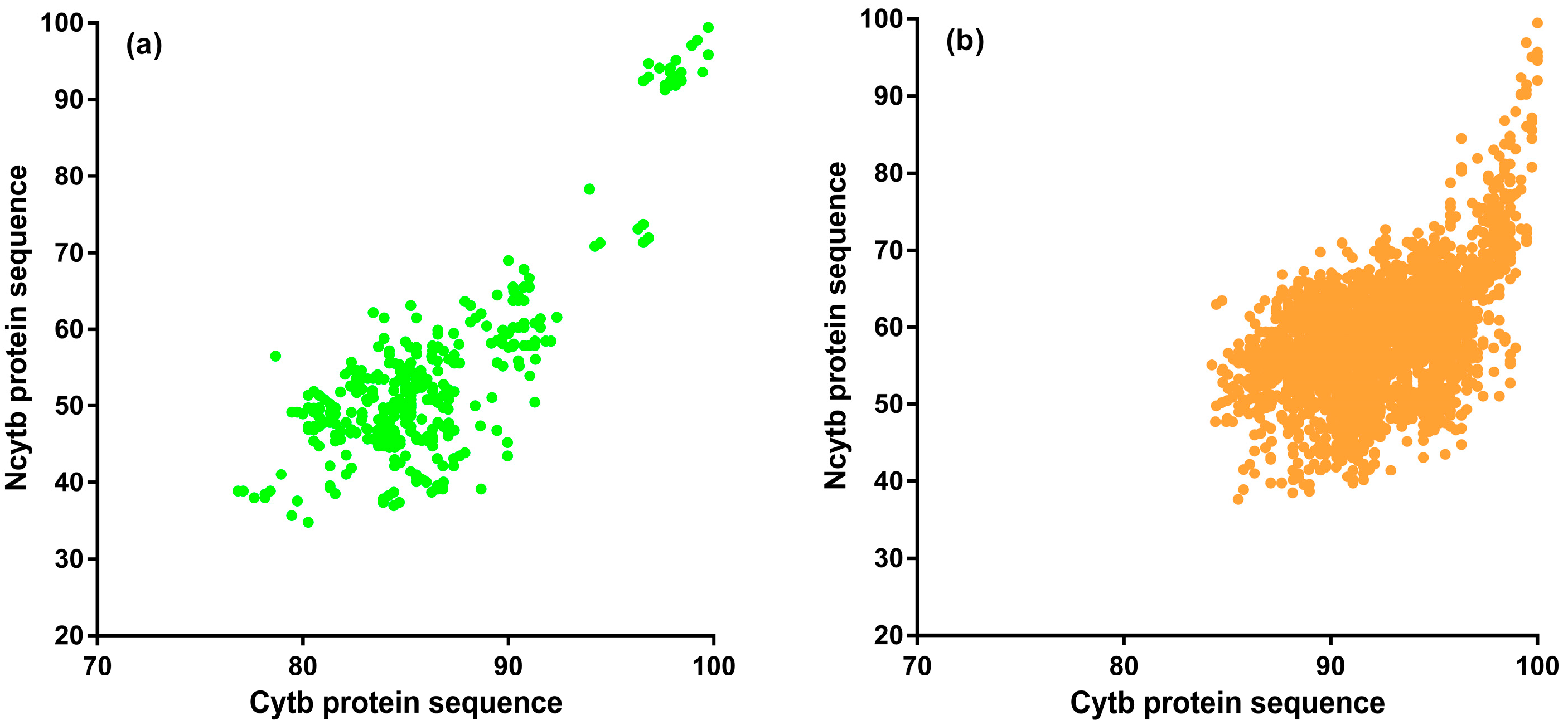
3.2. Sequence Homology of Putative ncytb-Encoded Proteins
3.3. Amino Acid Conservation of ncytb-Encoded Proteins
3.4. Predicted Properties of ncytb-Encoded Proteins
3.5. Cytosolic Integrity of the cytb Primary Open Reading Frame
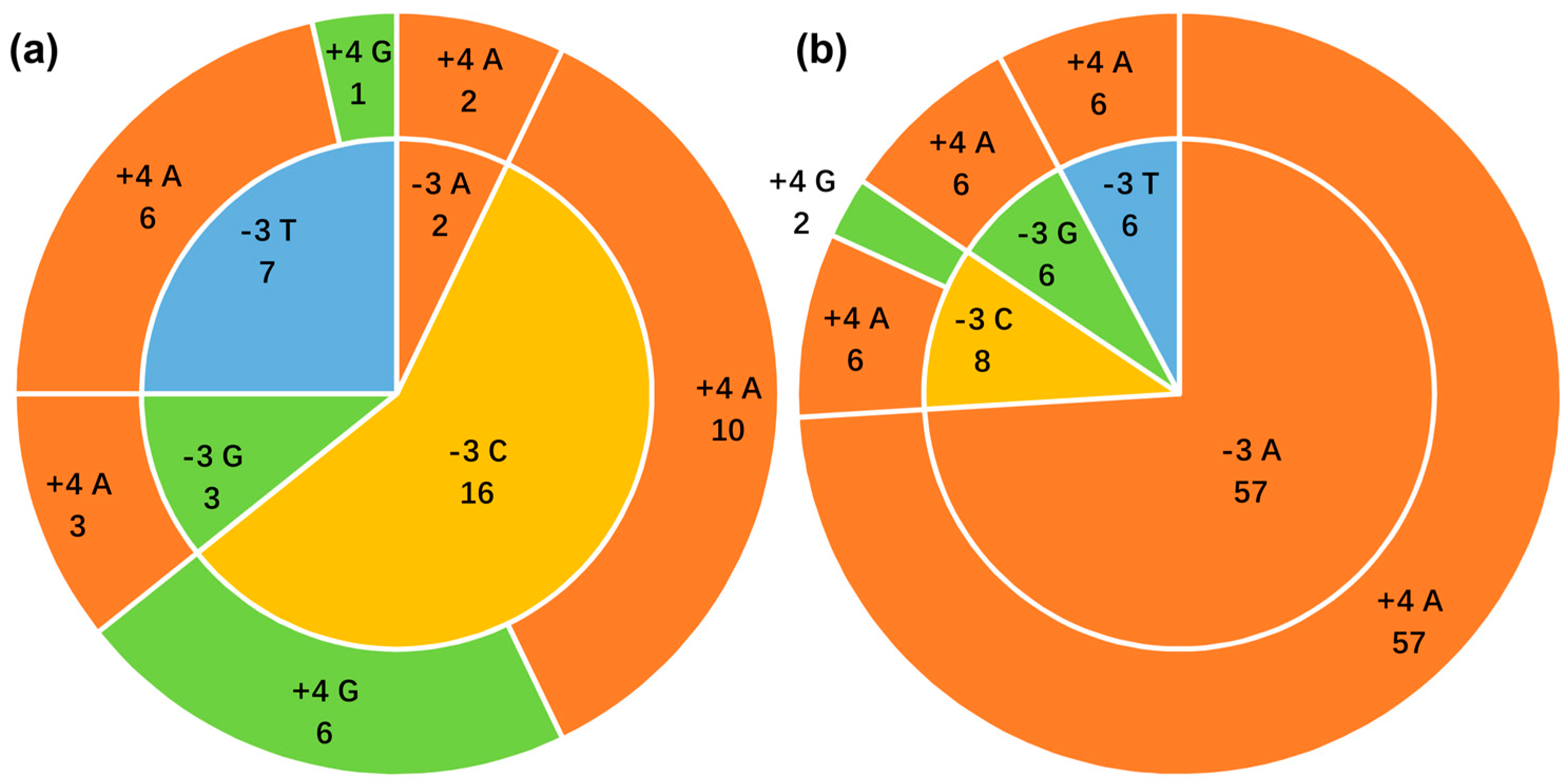
3.6. Context of the ncytb AUG Initiation Codon
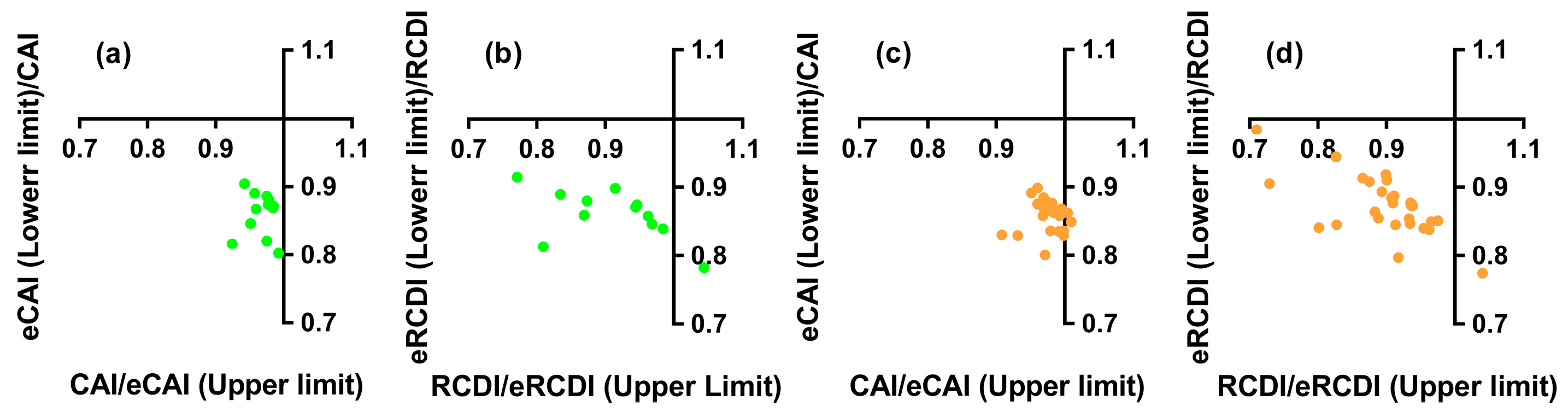
3.7. Codon Adaptation and Deoptimization of the Nested ncytb Gene
3.8. Codon Usage Similarity of the Nested ncytb Gene
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AltORF | alternative open reading frame |
| BLASTp | protein-protein BLAST |
| CAI | Codon Adaptation Index |
| CDS | coding sequences |
| CoCoPUTs | Codon and Codon-Pair Usage Tables |
| cytb | cytochrome b |
| Cytb | Cytochrome b |
| eCAI | expected CAI |
| eRCDI | expected RCDI |
| nAltORF | nested alternative open reading frame |
| ncytb | nested ORF of cytb |
| Ncytb | Protein of ncytb gene |
| pI | isoelectric point |
| PTC | premature termination codons |
| RCDI | Relative Codon Deoptimization Index |
| SMS | Sequence Manipulation Suite |
References
- Vanderperre, B.; Lucier, J.F.; Bissonnette, C.; Motard, J.; Tremblay, G.; Vanderperre, S.; Wisztorski, M.; Salzet, M.; Boisvert, F.M.; Roucou, X. Direct detection of alternative open reading frames translation products in human significantly expands the proteome. PLoS ONE 2013, 8, e70698. [Google Scholar] [CrossRef] [PubMed]
- Vasu, K.; Khan, D.; Ramachandiran, I.; Blankenberg, D.; Fox, P.L. Analysis of nested alternate open reading frames and their encoded proteins. NAR Genom. Bioinform. 2022, 4, lqac076. [Google Scholar] [CrossRef]
- Watanabe, K. Unique features of animal mitochondrial translation systems. The non-universal genetic code, unusual features of the translational apparatus and their relevance to human mitochondrial diseases. Proc. Jpn. Academy. Ser. B Phys. Biol. Sci. 2010, 86, 11–39. [Google Scholar] [CrossRef]
- Rackham, O.; Filipovska, A. Organization and expression of the mammalian mitochondrial genome. Nat. Rev. Genet. 2022, 23, 606–623. [Google Scholar] [CrossRef]
- Hu, Z.; Yang, L.; Zhang, M.; Tang, H.; Huang, Y.; Su, Y.; Ding, Y.; Li, C.; Wang, M.; Zhou, Y.; et al. A novel protein CYTB-187AA encoded by the mitochondrial gene CYTB modulates mammalian early development. Cell Metab. 2024, 36, 1586–1597.e7. [Google Scholar] [CrossRef]
- Janssen, K.A.; Song, A. From whence it came: Mitochondrial mRNA leaves, a protein returns. Cell Metab. 2024, 36, 1433–1435. [Google Scholar] [CrossRef]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon usage bias. Mol. Biol. Rep. 2022, 49, 539–565. [Google Scholar] [CrossRef]
- Alexaki, A.; Kames, J.; Holcomb, D.D.; Athey, J.; Santana-Quintero, L.V.; Lam, P.V.N.; Hamasaki-Katagiri, N.; Osipova, E.; Simonyan, V.; Bar, H.; et al. Codon and Codon-Pair Usage Tables (CoCoPUTs): Facilitating Genetic Variation Analyses and Recombinant Gene Design. J. Mol. Biol. 2019, 431, 2434–2441. [Google Scholar] [CrossRef]
- Stothard, P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. BioTechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Nicholas, K.B.; Nicholas, H.B.J. GeneDoc: A Tool for Editing and Annotating Multiple Sequence Alignments. 1997. Available online: http://www.nrbsc.org/gfx/genedoc/ (accessed on 23 June 2025).
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Ødum, M.T.; Teufel, F.; Thumuluri, V.; Almagro Armenteros, J.J.; Johansen, A.R.; Winther, O.; Nielsen, H. DeepLoc 2.1: Multi-label membrane protein type prediction using protein language models. Nucleic Acids Res. 2024, 52, W215–W220. [Google Scholar] [CrossRef] [PubMed]
- Hernández, G.; Osnaya, V.G.; Pérez-Martínez, X. Conservation and Variability of the AUG Initiation Codon Context in Eukaryotes. Trends Biochem. Sci. 2019, 44, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, Y.; Tsuji, J.; Fu, S.C.; Tomii, K.; Horton, P.; Imai, K. MitoFates: Improved prediction of mitochondrial targeting sequences and their cleavage sites. Mol. Cell. Proteom. 2015, 14, 1113–1126. [Google Scholar] [CrossRef] [PubMed]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Almagro Armenteros, J.J.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. bioRxiv 2022. bioRxiv: 2022.04.08.487609. [Google Scholar] [CrossRef]
- Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; et al. The conserved domain database in 2023. Nucleic Acids Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef]
- Kozlowski, L.P. IPC 2.0: Prediction of isoelectric point and pKa dissociation constants. Nucleic Acids Res. 2021, 49, W285–W292. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. The codon Adaptation Index--a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- Xia, X. An improved implementation of codon adaptation index. Evol. Bioinform. Online 2007, 3, 53–58. [Google Scholar] [CrossRef]
- Puigbò, P.; Bravo, I.G.; Garcia-Vallvé, S. E-CAI: A novel server to estimate an expected value of Codon Adaptation Index (eCAI). BMC Bioinform. 2008, 9, 65. [Google Scholar] [CrossRef]
- Mueller, S.; Papamichail, D.; Coleman, J.R.; Skiena, S.; Wimmer, E. Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J. Virol. 2006, 80, 9687–9696. [Google Scholar] [CrossRef] [PubMed]
- Puigbò, P.; Aragonès, L.; Garcia-Vallvé, S. RCDI/eRCDI: A web-server to estimate codon usage deoptimization. BMC Res. Notes 2010, 3, 87. [Google Scholar] [CrossRef] [PubMed]
- Silverj, A.; Rota-Stabelli, O. On the correct interpretation of similarity index in codon usage studies: Comparison with four other metrics and implications for Zika and West Nile virus. Virus Res. 2020, 286, 198097. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.L.; Xia, R.X. Codon Usage in the Iflaviridae Family Is Not Diverse Though the Family Members Are Isolated from Diverse Host Taxa. Viruses 2019, 11, 1087. [Google Scholar] [CrossRef]
- Orr, M.W.; Mao, Y.; Storz, G.; Qian, S.B. Alternative ORFs and small ORFs: Shedding light on the dark proteome. Nucleic Acids Res. 2020, 48, 1029–1042. [Google Scholar] [CrossRef]
- Mouilleron, H.; Delcourt, V.; Roucou, X. Death of a dogma: Eukaryotic mRNAs can code for more than one protein. Nucleic Acids Res. 2016, 44, 14–23. [Google Scholar] [CrossRef]
- Korona, R. Gene dispensability. Curr. Opin. Biotechnol. 2011, 22, 547–551. [Google Scholar] [CrossRef]
- Yocca, A.E.; Edger, P.P. Machine learning approaches to identify core and dispensable genes in pangenomes. Plant Genome 2022, 15, e20135. [Google Scholar] [CrossRef]
- Addou, S.; Rentzsch, R.; Lee, D.; Orengo, C.A. Domain-based and family-specific sequence identity thresholds increase the levels of reliable protein function transfer. J. Mol. Biol. 2009, 387, 416–430. [Google Scholar] [CrossRef]
- Ladunga, I. Finding Homologs in Amino Acid Sequences Using Network BLAST Searches. Curr. Protoc. Bioinform. 2017, 59, 3.4.1–3.4.24. [Google Scholar] [CrossRef]
- Golicz, A.A.; Bayer, P.E.; Bhalla, P.L.; Batley, J.; Edwards, D. Pangenomics Comes of Age: From Bacteria to Plant and Animal Applications. Trends Genet. 2020, 36, 132–145. [Google Scholar] [CrossRef]
- Xie, J.; Zhuang, Z.; Gou, S.; Zhang, Q.; Wang, X.; Lan, T.; Lian, M.; Li, N.; Liang, Y.; Ouyang, Z.; et al. Precise genome editing of the Kozak sequence enables bidirectional and quantitative modulation of protein translation to anticipated levels without affecting transcription. Nucleic Acids Res. 2023, 51, 10075–10093. [Google Scholar] [CrossRef]
- Novoa, E.M.; Ribas de Pouplana, L. Speeding with control: Codon usage, tRNAs, and ribosomes. Trends Genet. 2012, 28, 574–581. [Google Scholar] [CrossRef]
- Davyt, M.; Bharti, N.; Ignatova, Z. Effect of mRNA/tRNA mutations on translation speed: Implications for human diseases. J. Biol. Chem. 2023, 299, 105089. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, S.-L.; Li, D.-T.; Liu, Y.-Q. Patchy Phylogenetic Distribution and Poor Translational Adaptation of a Nested ORF in the Mammalian Mitochondrial cytb Gene. Genes 2025, 16, 833. https://doi.org/10.3390/genes16070833
Shi S-L, Li D-T, Liu Y-Q. Patchy Phylogenetic Distribution and Poor Translational Adaptation of a Nested ORF in the Mammalian Mitochondrial cytb Gene. Genes. 2025; 16(7):833. https://doi.org/10.3390/genes16070833
Chicago/Turabian StyleShi, Sheng-Lin, Dan-Tong Li, and Yan-Qun Liu. 2025. "Patchy Phylogenetic Distribution and Poor Translational Adaptation of a Nested ORF in the Mammalian Mitochondrial cytb Gene" Genes 16, no. 7: 833. https://doi.org/10.3390/genes16070833
APA StyleShi, S.-L., Li, D.-T., & Liu, Y.-Q. (2025). Patchy Phylogenetic Distribution and Poor Translational Adaptation of a Nested ORF in the Mammalian Mitochondrial cytb Gene. Genes, 16(7), 833. https://doi.org/10.3390/genes16070833