
Epigenetics Plays a Role in the Pathogenesis and Treatment of Diabetes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Introduction to Diabetes
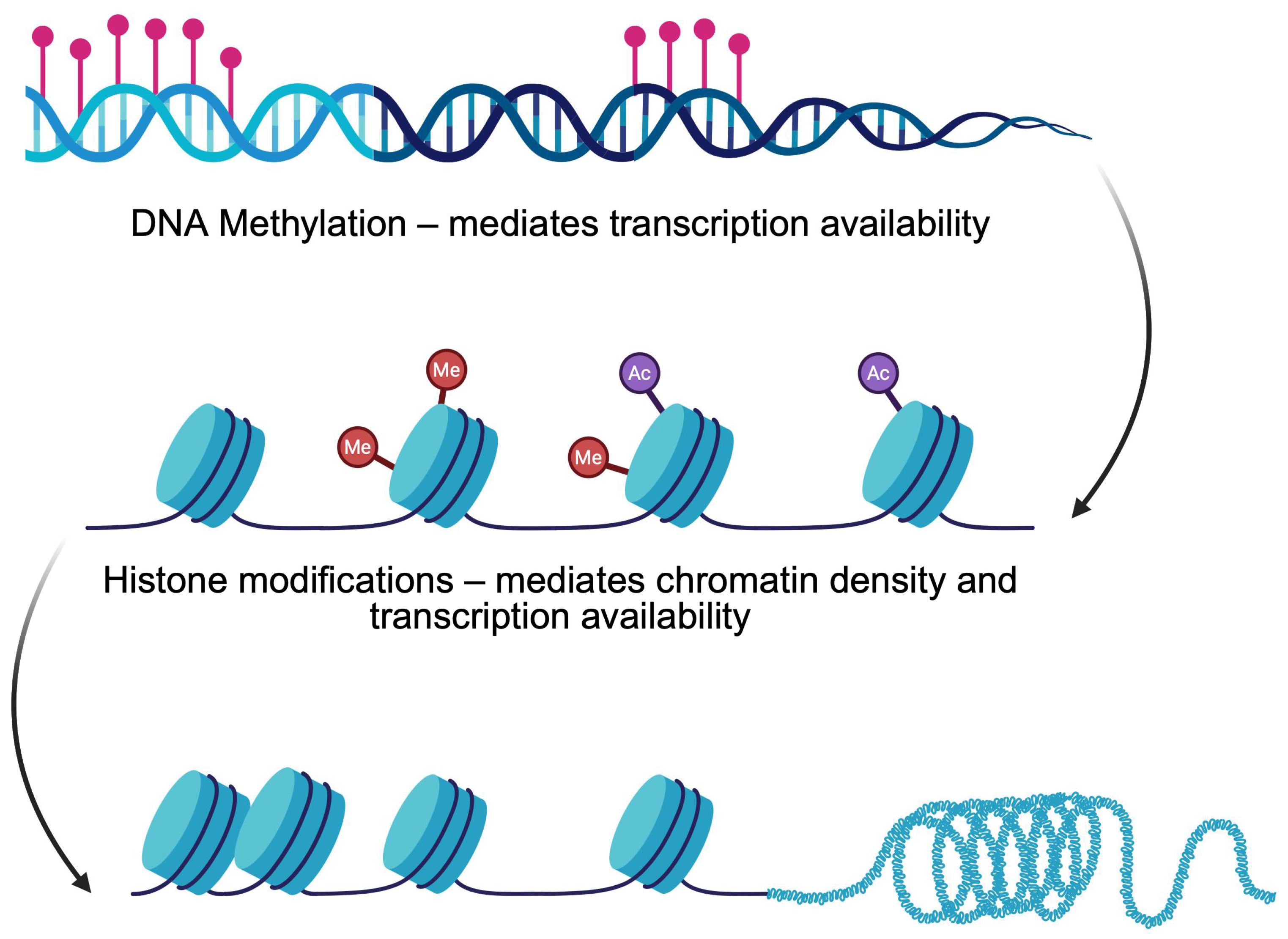
1.2. Introduction to Epigenetics
1.3. Methodology
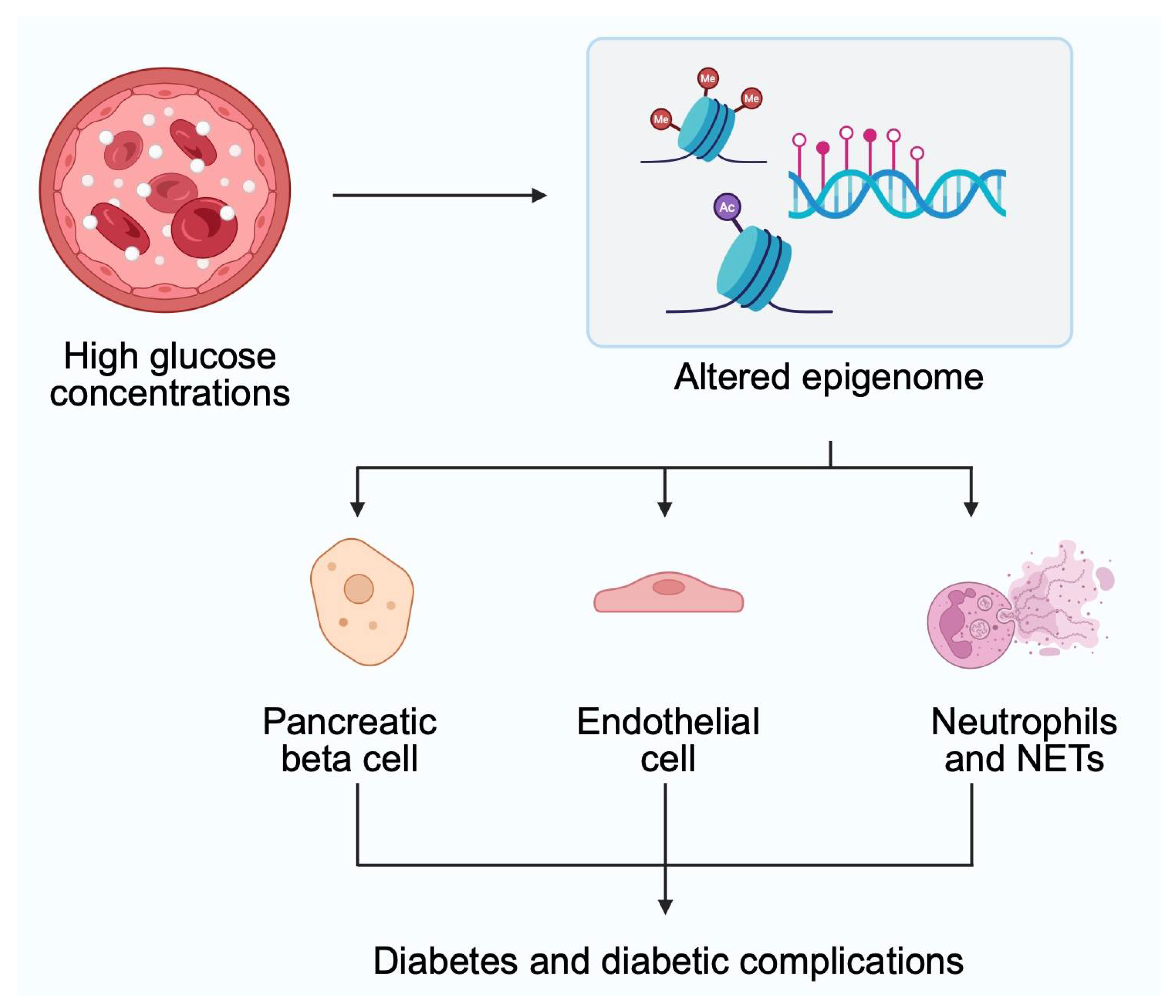
2. Epigenetics and Diabetes
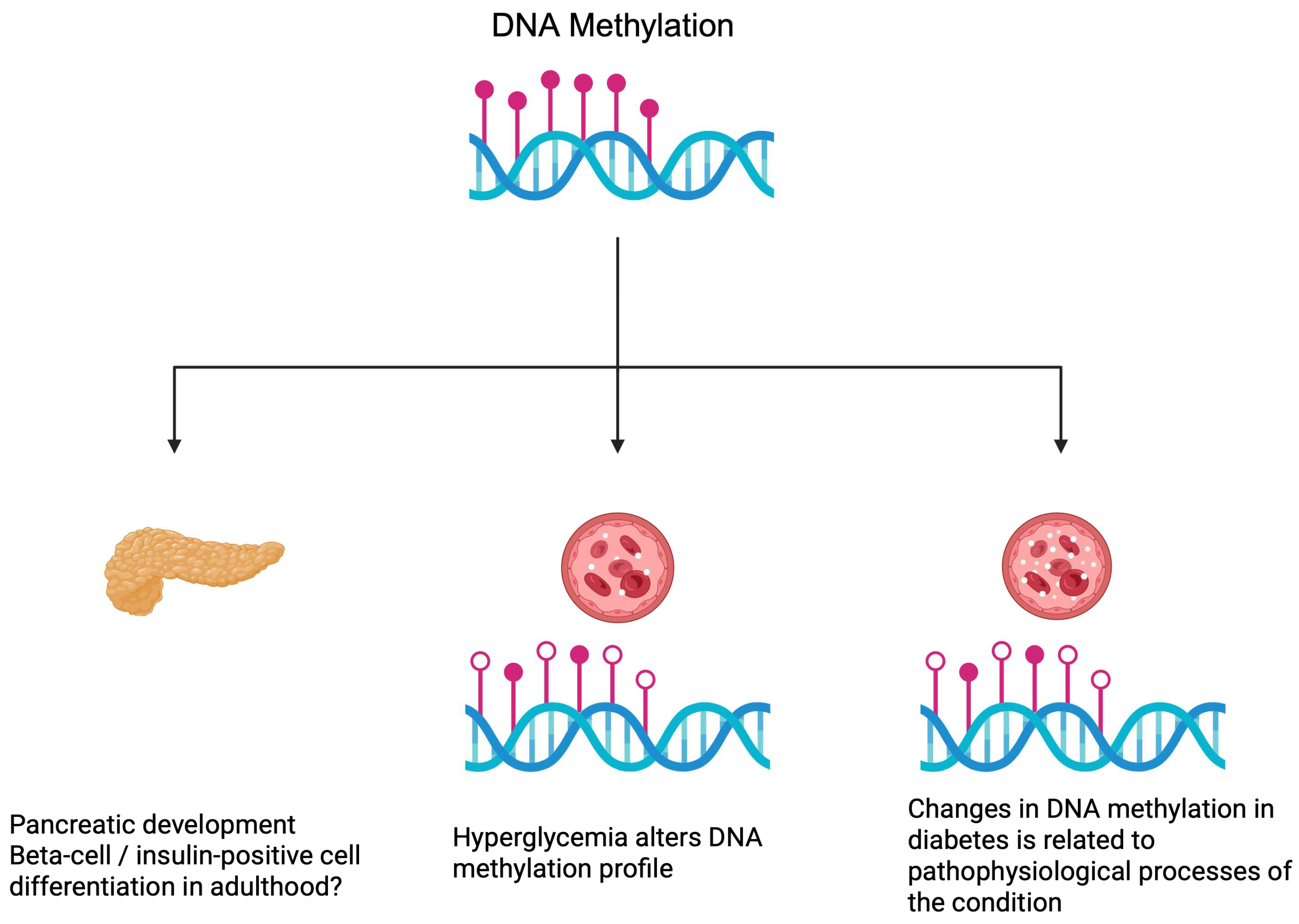
2.1. DNA Methylation
2.2. Histone Modifications
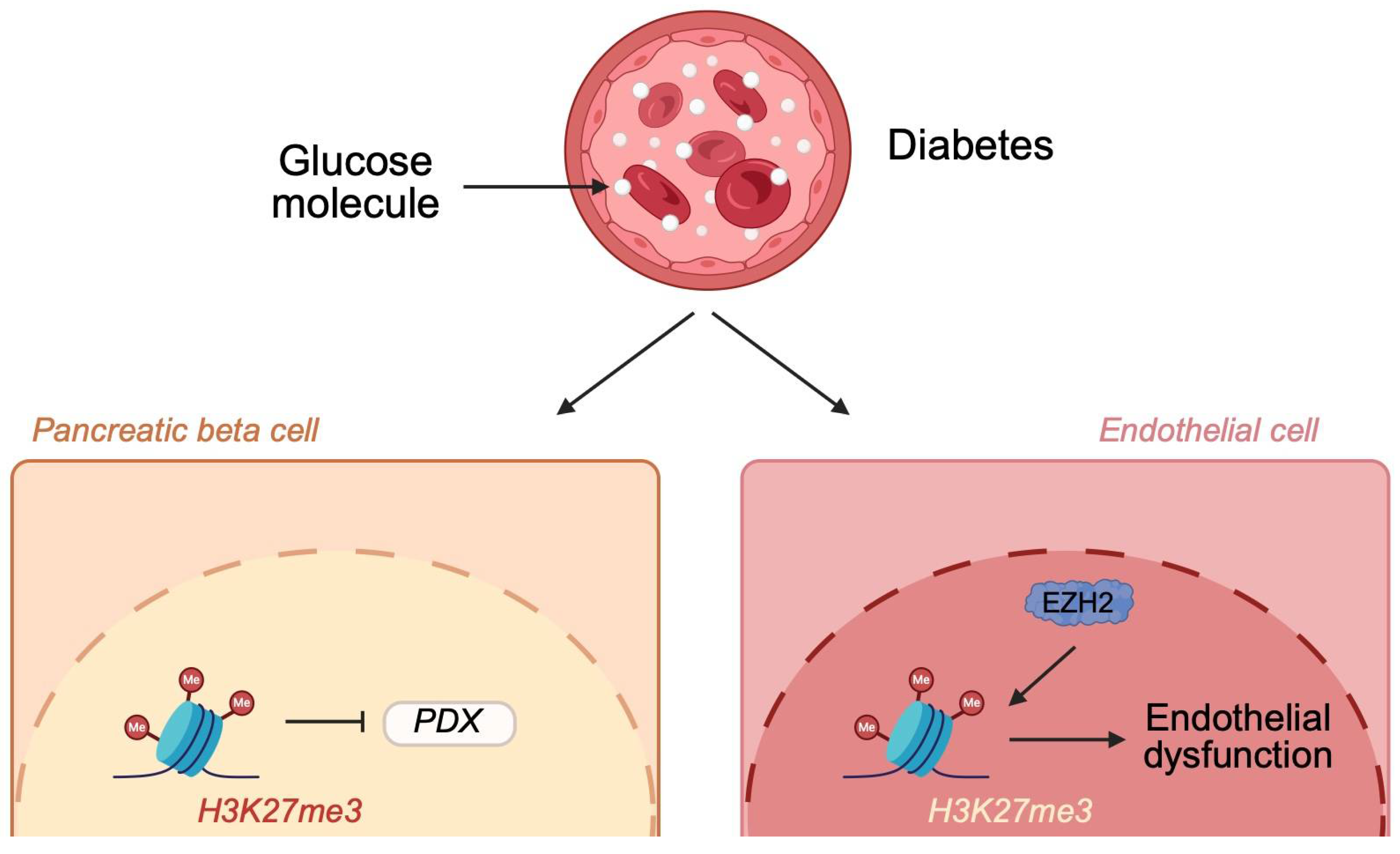
2.2.1. Histone Methylation
2.2.2. Histone Acetylation
2.2.3. Histone Lactylation
3. Limitations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DM | Diabetes mellitus |
| T1DM | Type 1 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| ADK | Adenosine kinase |
| TH | Tyrosine hydroxylase |
| TXNIP | Thioredoxin-interacting protein |
| NAFLD | Non-alcoholic fatty liver disease |
| EZH2 | Enhancer of zeste homolog 2 |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| HATs | Histone acetyltransferases |
| HDAC | Histone deacetylase |
| NLRP3 | NOD-like receptor (NLR) family pyrin domain-containing 3 |
References
- Accili, D.; Deng, Z.; Liu, Q. Insulin resistance in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2025, 21, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Bonnefond, A.; Unnikrishnan, R.; Doria, A.; Vaxillaire, M.; Kulkarni, R.N.; Mohan, V.; Trischitta, V.; Froguel, P. Monogenic diabetes. Nat. Rev. Dis. Primers 2023, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association Professional Practice Committee. 9. Pharmacologic Approaches to Glycemic Treatment: Standards of Care in Diabetes-2025. Diabetes Care 2025, 48, S181–S206. [Google Scholar] [CrossRef]
- Drucker, D.J. GLP-1-based therapies for diabetes, obesity and beyond. Nat. Rev. Drug Discov. 2025. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Scisciola, L.; Rizzo, M.R.; Cataldo, V.; Fontanella, R.A.; Balestrieri, M.L.; D’Onofrio, N.; Marfella, R.; Paolisso, G.; Barbieri, M. Incretin drugs effect on epigenetic machinery: New potential therapeutic implications in preventing vascular diabetic complications. FASEB J. 2020, 34, 16489–16503. [Google Scholar] [CrossRef]
- Smith, Z.D.; Hetzel, S.; Meissner, A. DNA methylation in mammalian development and disease. Nat. Rev. Genet. 2025, 26, 7–30. [Google Scholar] [CrossRef]
- Yousefi, P.D.; Suderman, M.; Langdon, R.; Whitehurst, O.; Davey Smith, G.; Relton, C.L. DNA methylation-based predictors of health: Applications and statistical considerations. Nat. Rev. Genet. 2022, 23, 369–383. [Google Scholar] [CrossRef]
- Yokoi, A.; Asahara, S.I.; Inoue, H.; Seike, M.; Kido, N.; Suzuki, H.; Kanno, A.; Kimura-Koyanagi, M.; Kido, Y.; Ogawa, W. Dapagliflozin administration to a mouse model of type 2 diabetes induces DNA methylation and gene expression changes in pancreatic islets. Biochem. Biophys. Res. Commun. 2024, 725, 150254. [Google Scholar] [CrossRef]
- Lai, L.; Juntilla, D.L.; Del C Gomez-Alonso, M.; Grallert, H.; Thorand, B.; Farzeen, A.; Rathmann, W.; Winkelmann, J.; Prokisch, H.; Gieger, C.; et al. Longitudinal association between DNA methylation and type 2 diabetes: Findings from the KORA F4/FF4 study. Cardiovasc. Diabetol. 2025, 24, 19. [Google Scholar] [CrossRef]
- Miller, R.G.; Mychaleckyj, J.C.; Onengut-Gumuscu, S.; Orchard, T.J.; Costacou, T. An Epigenome-Wide Association Study of DNA Methylation and Proliferative Retinopathy over 28 Years in Type 1 Diabetes. Ophthalmol. Sci. 2024, 4, 100497. [Google Scholar] [CrossRef]
- Parveen, N.; Wang, J.K.; Bhattacharya, S.; Cuala, J.; Rajkumar, M.S.; Butler, A.E.; Wu, X.; Shih, H.P.; Georgia, S.K.; Dhawan, S. DNA Methylation-Dependent Restriction of Tyrosine Hydroxylase Contributes to Pancreatic beta-Cell Heterogeneity. Diabetes 2023, 72, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Takagi, H.; Kuno, M.; Sasaki, T.; Taki, K.; Ito, Y.; Miyata, T.; Kobayashi, T.; Sugiyama, M.; Onoue, T.; et al. Dapagliflozin increased pancreatic beta cell proliferation and insulinogenic index in mice fed a high-fat and high-sodium chloride diet. Biochem. Biophys. Res. Commun. 2025, 749, 151364. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Yu, J.; Guo, X.; Wang, S.; Wang, Z.; Chen, Y.; Hu, X.; Li, H.; Chen, L.; Zheng, J. Adenosine kinase inhibits beta-cell proliferation by upregulating DNA methyltransferase 3A expression. Diabetes Obes. Metab. 2024, 26, 2956–2968. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.C.H.; Huang, W.; Fong, S.W.; Chan, C.; Lee, K.C.; Yeung, W.S.B.; Lee, Y.L. Hyperglycemia Altered DNA Methylation Status and Impaired Pancreatic Differentiation from Embryonic Stem Cells. Int. J. Mol. Sci. 2021, 22, 10729. [Google Scholar] [CrossRef]
- Blotsky, A.L.; Rahme, E.; Dahhou, M.; Nakhla, M.; Dasgupta, K. Gestational diabetes associated with incident diabetes in childhood and youth: A retrospective cohort study. CMAJ 2019, 191, E410–E417. [Google Scholar] [CrossRef]
- Guo, H.Y.; Tang, S.B.; Li, L.J.; Lin, J.; Zhang, T.T.; Chao, S.; Jin, X.W.; Xu, K.P.; Su, X.F.; Yin, S.; et al. Gestational diabetes mellitus causes genome hyper-methylation of oocyte via increased EZH2. Nat. Commun. 2025, 16, 127. [Google Scholar] [CrossRef]
- Qadir, M.M.F.; Alvarez-Cubela, S.; Klein, D.; van Dijk, J.; Muniz-Anquela, R.; Moreno-Hernandez, Y.B.; Lanzoni, G.; Sadiq, S.; Navarro-Rubio, B.; Garcia, M.T.; et al. Single-cell resolution analysis of the human pancreatic ductal progenitor cell niche. Proc. Natl. Acad. Sci. USA 2020, 117, 10876–10887. [Google Scholar] [CrossRef]
- Zhao, H.; Huang, X.; Liu, Z.; Pu, W.; Lv, Z.; He, L.; Li, Y.; Zhou, Q.; Lui, K.O.; Zhou, B. Pre-existing beta cells but not progenitors contribute to new beta cells in the adult pancreas. Nat. Metab. 2021, 3, 352–365. [Google Scholar] [CrossRef]
- Hatano, R.; Zhang, X.; Lee, E.; Kaneda, A.; Tanaka, T.; Miki, T. Mosaic ablation of pancreatic beta cells induces de-differentiation and repetitive proliferation of residual beta cells in adult mice. iScience 2024, 27, 110656. [Google Scholar] [CrossRef]
- Alhazzaa, R.A.; McKinley, R.E.; Getachew, B.; Tizabi, Y.; Heinbockel, T.; Csoka, A.B. Epigenetic Changes Induced by High Glucose in Human Pancreatic Beta Cells. J. Diabetes Res. 2023, 2023, 9947294. [Google Scholar] [CrossRef]
- Marumo, T.; Yoshida, N.; Inoue, N.; Yamanouchi, M.; Ubara, Y.; Urakami, S.; Fujii, T.; Takazawa, Y.; Ohashi, K.; Kawarazaki, W.; et al. Aberrant proximal tubule DNA methylation underlies phenotypic changes related to kidney dysfunction in patients with diabetes. Am. J. Physiol. Renal Physiol. 2024, 327, F397–F411. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, W.; Zhu, Y.; Zhang, S.; Jiang, M.; Hu, J.; Zhang, H.H. Genomic DNA Methylation in Diabetic Chronic Complications in Patients with Type 2 Diabetes Mellitus. Front. Endocrinol. 2022, 13, 896511. [Google Scholar] [CrossRef] [PubMed]
- Fraszczyk, E.; Thio, C.H.L.; Wackers, P.; Dolle, M.E.T.; Bloks, V.W.; Hodemaekers, H.; Picavet, H.S.; Stynenbosch, M.; Verschuren, W.M.M.; Snieder, H.; et al. DNA methylation trajectories and accelerated epigenetic aging in incident type 2 diabetes. Geroscience 2022, 44, 2671–2684. [Google Scholar] [CrossRef] [PubMed]
- Juvinao-Quintero, D.L.; Sharp, G.C.; Sanderson, E.C.M.; Relton, C.L.; Elliott, H.R. Investigating causality in the association between DNA methylation and type 2 diabetes using bidirectional two-sample Mendelian randomisation. Diabetologia 2023, 66, 1247–1259. [Google Scholar] [CrossRef]
- Carry, P.M.; Vanderlinden, L.A.; Johnson, R.K.; Buckner, T.; Steck, A.K.; Kechris, K.; Yang, I.V.; Fingerlin, T.E.; Fiehn, O.; Rewers, M.; et al. Longitudinal changes in DNA methylation during the onset of islet autoimmunity differentiate between reversion versus progression of islet autoimmunity. Front. Immunol. 2024, 15, 1345494. [Google Scholar] [CrossRef]
- Wang, Z.; Peng, H.; Gao, W.; Cao, W.; Lv, J.; Yu, C.; Huang, T.; Sun, D.; Wang, B.; Liao, C.; et al. Blood DNA methylation markers associated with type 2 diabetes, fasting glucose, and HbA1c levels: An epigenome-wide association study in 316 adult twin pairs. Genomics 2021, 113, 4206–4213. [Google Scholar] [CrossRef]
- Xiang, Y.; Wang, Z.; Hui, Q.; Gwinn, M.; Vaccarino, V.; Sun, Y.V. DNA Methylation of TXNIP Independently Associated with Inflammation and Diabetes Mellitus in Twins. Twin Res. Hum. Genet. 2021, 24, 273–280. [Google Scholar] [CrossRef]
- Miller, R.G.; Mychaleckyj, J.C.; Onengut-Gumuscu, S.; Orchard, T.J.; Costacou, T. TXNIP DNA methylation is associated with glycemic control over 28 years in type 1 diabetes: Findings from the Pittsburgh Epidemiology of Diabetes Complications (EDC) study. BMJ Open Diabetes Res. Care 2023, 11, e003068. [Google Scholar] [CrossRef]
- Schneider, M.R.; Zettler, S.; Rathkolb, B.; Dahlhoff, M. TXNIP overexpression in mice enhances streptozotocin-induced diabetes severity. Mol. Cell. Endocrinol. 2023, 565, 111885. [Google Scholar] [CrossRef]
- Schroder, B.; Kahl, S.; Roden, M. Non-alcoholic fatty liver disease in type 2 diabetes—A specific entity? Liver Int. 2021, 41 (Suppl. S1), 105–111. [Google Scholar] [CrossRef]
- Guo, Y.; Chen, J.; Liu, N.; Liu, Z.; Shi, B.; Sun, H. Association of Circulating TXNIP Levels with Fatty Liver in Newly Diagnosed Type 2 Diabetes Mellitus. Diabetes Metab. Syndr. Obes. 2022, 15, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Amin, F.M.; Abdelaziz, R.R.; Hamed, M.F.; Nader, M.A.; Shehatou, G.S.G. Dimethyl fumarate ameliorates diabetes-associated vascular complications through ROS-TXNIP-NLRP3 inflammasome pathway. Life Sci. 2020, 256, 117887. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Wu, M.; Song, S.; Bian, Y.; Shi, Y. TXNIP deficiency attenuates renal fibrosis by modulating mTORC1/TFEB-mediated autophagy in diabetic kidney disease. Ren. Fail. 2024, 46, 2338933. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Guan, L.; Su, W.; Zhao, N.; Song, X.; Wang, J.; Tang, X.; Li, W.; Jiao, X. TXNIP inhibition in the treatment of type 2 diabetes mellitus: Design, synthesis, and biological evaluation of quinazoline derivatives. J. Enzym. Inhib. Med. Chem. 2023, 38, 2166937. [Google Scholar] [CrossRef]
- Vojta, A.; Dobrinic, P.; Tadic, V.; Bockor, L.; Korac, P.; Julg, B.; Klasic, M.; Zoldos, V. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef]
- Park, H.; Shin, J.; Kim, Y.; Saito, T.; Saido, T.C.; Kim, J. CRISPR/dCas9-Dnmt3a-mediated targeted DNA methylation of APP rescues brain pathology in a mouse model of Alzheimer’s disease. Transl. Neurodegener. 2022, 11, 41. [Google Scholar] [CrossRef]
- Zhu, M.; Huang, Q.; Li, H.; Zhao, Y.; Guo, H.; Wang, T.; Liu, X.; Huang, Y.; Hu, J.; Fang, C.; et al. The impact of site-specific DNA methylation in KCNJ11 promoter on type 2 diabetes. Heliyon 2024, 10, e39934. [Google Scholar] [CrossRef]
- Wu, B.; Xu, W. Case report: Neonatal diabetes mellitus caused by KCNJ11 mutation presenting with intracranial hemorrhage. Front. Neurol. 2023, 14, 1072078. [Google Scholar] [CrossRef]
- Song, X.; Cao, Y.; Ye, J.; Dai, W.; Zhang, S.; Ye, S. A new mutation c.685G>A:p.E229K in the KCNJ11 gene: A case report of maturity-onset diabetes of the young13. Medicine 2022, 101, e30668. [Google Scholar] [CrossRef]
- Karaglani, M.; Panagopoulou, M.; Cheimonidi, C.; Tsamardinos, I.; Maltezos, E.; Papanas, N.; Papazoglou, D.; Mastorakos, G.; Chatzaki, E. Liquid Biopsy in Type 2 Diabetes Mellitus Management: Building Specific Biosignatures via Machine Learning. J. Clin. Med. 2022, 11, 1045. [Google Scholar] [CrossRef]
- Mattei, A.L.; Bailly, N.; Meissner, A. DNA methylation: A historical perspective. Trends Genet. 2022, 38, 676–707. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhang, K.; Wang, Y.; Gu, Y. Comprehensive review of histone lactylation: Structure, function, and therapeutic targets. Biochem. Pharmacol. 2024, 225, 116331. [Google Scholar] [CrossRef]
- Padeken, J.; Methot, S.P.; Gasser, S.M. Establishment of H3K9-methylated heterochromatin and its functions in tissue differentiation and maintenance. Nat. Rev. Mol. Cell Biol. 2022, 23, 623–640. [Google Scholar] [CrossRef] [PubMed]
- Tu, P.; Huang, B.; Li, M.; Zhang, Y.; Bao, S.; Tu, N.; Yang, Y.; Lu, J. Exendin-4 may improve type 2 diabetes by modulating the epigenetic modifications of pancreatic histone H3 in STZ-induced diabetic C57BL/6 J mice. J. Physiol. Biochem. 2022, 78, 51–59. [Google Scholar] [CrossRef]
- Sanchez-Ceinos, J.; Hussain, S.; Khan, A.W.; Zhang, L.; Almahmeed, W.; Pernow, J.; Cosentino, F. Repressive H3K27me3 drives hyperglycemia-induced oxidative and inflammatory transcriptional programs in human endothelium. Cardiovasc. Diabetol. 2024, 23, 122. [Google Scholar] [CrossRef]
- Li, S.S.; Pan, L.; Zhang, Z.Y.; Zhou, M.D.; Chen, X.F.; Qian, L.L.; Dai, M.; Lu, J.; Yu, Z.M.; Dang, S.; et al. Diabetes Promotes Myocardial Fibrosis via AMPK/EZH2/PPAR-gamma Signaling Pathway. Diabetes Metab. J. 2024, 48, 716–729. [Google Scholar] [CrossRef]
- Al-Hasani, K.; Marikar, S.N.; Kaipananickal, H.; Maxwell, S.; Okabe, J.; Khurana, I.; Karagiannis, T.; Liang, J.J.; Mariana, L.; Loudovaris, T.; et al. EZH2 inhibitors promote beta-like cell regeneration in young and adult type 1 diabetes donors. Signal Transduct. Target. Ther. 2024, 9, 2. [Google Scholar] [CrossRef]
- Kojima, K.; Nakamura, N.; Hayashi, A.; Kondo, S.; Miyabe, M.; Kikuchi, T.; Sawada, N.; Saiki, T.; Minato, T.; Ozaki, R.; et al. Impacts of Hyperglycemia on Epigenetic Modifications in Human Gingival Fibroblasts and Gingiva in Diabetic Rats. Int. J. Mol. Sci. 2024, 25, 10979. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Radhakrishnan, R.; Mohammad, G. Regulation of Rac1 transcription by histone and DNA methylation in diabetic retinopathy. Sci. Rep. 2021, 11, 14097. [Google Scholar] [CrossRef]
- Alka, K.; Mohammad, G.; Kowluru, R.A. Regulation of serine palmitoyl-transferase and Rac1-Nox2 signaling in diabetic retinopathy. Sci. Rep. 2022, 12, 16740. [Google Scholar] [CrossRef]
- Tang, Y.; Hu, Y.; Ding, X.; Luo, D.; Li, C.; Daraqel, B.; Zheng, L. Enriched H3K27Me3 on BMP4 suppresses the osteoblastic differentiation potential of BMSCs in diabetes mellitus. Biochem. Biophys. Res. Commun. 2024, 735, 150741. [Google Scholar] [CrossRef] [PubMed]
- Charidemou, E.; Kirmizis, A. A two-way relationship between histone acetylation and metabolism. Trends Biochem. Sci. 2024, 49, 1046–1062. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.W.; Sheng, H.; Bai, Y.F.; Weng, Y.Y.; Fan, X.Y.; Zheng, F.; Fu, J.Q.; Zhang, F. Inhibition of histone acetyltransferase by naringenin and hesperetin suppresses Txnip expression and protects pancreatic beta cells in diabetic mice. Phytomedicine 2021, 88, 153454. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Lee, Y.B.; Lee, H.; Choi, Y.K.; Park, B.Y.; Kim, M.J.; Youn, Y.J.; Kim, S.H.; Jung, S.J.; Song, D.K.; et al. Diabetes Primes Neutrophils for Neutrophil Extracellular Trap Formation through Trained Immunity. Research 2024, 7, 0365. [Google Scholar] [CrossRef]
- Shafqat, A.; Abdul Rab, S.; Ammar, O.; Al Salameh, S.; Alkhudairi, A.; Kashir, J.; Alkattan, K.; Yaqinuddin, A. Emerging role of neutrophil extracellular traps in the complications of diabetes mellitus. Front. Med. 2022, 9, 995993. [Google Scholar] [CrossRef]
- Yu, X.; Wu, H.; Su, J.; Liu, X.; Liang, K.; Li, Q.; Yu, R.; Shao, X.; Wang, H.; Wang, Y.L.; et al. Acetyl-CoA metabolism maintains histone acetylation for syncytialization of human placental trophoblast stem cells. Cell Stem Cell 2024, 31, 1280–1297.e7. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, F.; Tian, Y.; Xu, W.; Zhu, Q.; Li, Z.; Qiu, L.; Lu, X.; Peng, B.; Liu, X.; et al. PARylated PDHE1alpha generates acetyl-CoA for local chromatin acetylation and DNA damage repair. Nat. Struct. Mol. Biol. 2023, 30, 1719–1734. [Google Scholar] [CrossRef]
- Li, W.; Long, Q.; Wu, H.; Zhou, Y.; Duan, L.; Yuan, H.; Ding, Y.; Huang, Y.; Wu, Y.; Huang, J.; et al. Nuclear localization of mitochondrial TCA cycle enzymes modulates pluripotency via histone acetylation. Nat. Commun. 2022, 13, 7414. [Google Scholar] [CrossRef]
- Lu, J.; Li, X.Q.; Chen, P.P.; Zhang, J.X.; Liu, L.; Wang, G.H.; Liu, X.Q.; Jiang, T.T.; Wang, M.Y.; Liu, W.T.; et al. Activation of acetyl-CoA synthetase 2 mediates kidney injury in diabetic nephropathy. JCI Insight 2023, 8, e165817. [Google Scholar] [CrossRef]
- Lu, J.; Li, X.Q.; Chen, P.P.; Zhang, J.X.; Li, L.; Wang, G.H.; Liu, X.Q.; Jiang, C.M.; Ma, K.L. Acetyl-CoA synthetase 2 promotes diabetic renal tubular injury in mice by rewiring fatty acid metabolism through SIRT1/ChREBP pathway. Acta Pharmacol. Sin. 2024, 45, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Zhen, J.; Sheng, X.; Chen, T.; Yu, H. Histone acetyltransferase Kat2a regulates ferroptosis via enhancing Tfrc and Hmox1 expression in diabetic cardiomyopathy. Cell Death Dis. 2024, 15, 406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, Y.; Ding, Y.; Jiang, Y.; Chen, H.; Zhan, Z.; Liu, X. Targeting KAT2A inhibits inflammatory macrophage activation and rheumatoid arthritis through epigenetic and metabolic reprogramming. MedComm 2023, 4, e306. [Google Scholar] [CrossRef] [PubMed]
- Lazar, A.G.; Vlad, M.L.; Manea, A.; Simionescu, M.; Manea, S.A. Activated Histone Acetyltransferase p300/CBP-Related Signalling Pathways Mediate Up-Regulation of NADPH Oxidase, Inflammation, and Fibrosis in Diabetic Kidney. Antioxidants 2021, 10, 1356. [Google Scholar] [CrossRef]
- Fan, Z.; Wu, J.; Chen, Q.N.; Lyu, A.K.; Chen, J.L.; Sun, Y.; Lyu, Q.; Zhao, Y.X.; Guo, A.; Liao, Z.Y.; et al. Type 2 diabetes-induced overactivation of P300 contributes to skeletal muscle atrophy by inhibiting autophagic flux. Life Sci. 2020, 258, 118243. [Google Scholar] [CrossRef]
- Kielbowski, K.; Szwedkowicz, A.; Plewa, P.; Bakinowska, E.; Becht, R.; Pawlik, A. Anticancer properties of histone deacetylase inhibitors—What is their potential? Expert Rev. Anticancer Ther. 2025, 25, 105–120. [Google Scholar] [CrossRef]
- Zhang, H.; D’Alessandro, A.; Li, M.; Reisz, J.A.; Riddle, S.; Muralidhar, A.; Bull, T.; Zhao, L.; Gerasimovskaya, E.; Stenmark, K.R. Histone deacetylase inhibitors synergize with sildenafil to suppress purine metabolism and proliferation in pulmonary hypertension. Vasc. Pharmacol. 2023, 149, 107157. [Google Scholar] [CrossRef]
- Theodoropoulou, M.A.; Mantzourani, C.; Kokotos, G. Histone Deacetylase (HDAC) Inhibitors as a Novel Therapeutic Option Against Fibrotic and Inflammatory Diseases. Biomolecules 2024, 14, 1605. [Google Scholar] [CrossRef]
- Guan, D.; Men, Y.; Bartlett, A.; Hernandez, M.A.S.; Xu, J.; Yi, X.; Li, H.S.; Kong, D.; Mazitschek, R.; Ozcan, U. Central inhibition of HDAC6 re-sensitizes leptin signaling during obesity to induce profound weight loss. Cell Metab. 2024, 36, 857–876.e10. [Google Scholar] [CrossRef]
- Vilarino-Garcia, T.; Polonio-Gonzalez, M.L.; Perez-Perez, A.; Ribalta, J.; Arrieta, F.; Aguilar, M.; Obaya, J.C.; Gimeno-Orna, J.A.; Iglesias, P.; Navarro, J.; et al. Role of Leptin in Obesity, Cardiovascular Disease, and Type 2 Diabetes. Int. J. Mol. Sci. 2024, 25, 2338. [Google Scholar] [CrossRef]
- Hou, Q.; Kan, S.; Wang, Z.; Shi, J.; Zeng, C.; Yang, D.; Jiang, S.; Liu, Z. Inhibition of HDAC6 with CAY10603 Ameliorates Diabetic Kidney Disease by Suppressing NLRP3 Inflammasome. Front. Pharmacol. 2022, 13, 938391. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xiao, G.Y.; Guo, T.; Song, Y.J.; Li, Q.M. Potential therapeutic role of pyroptosis mediated by the NLRP3 inflammasome in type 2 diabetes and its complications. Front. Endocrinol. 2022, 13, 986565. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Che, G.; Yang, Y.; Xie, H.; Tian, J. Histone Deacetylase 3 Aggravates Type 1 Diabetes Mellitus by Inhibiting Lymphocyte Apoptosis Through the microRNA-296-5p/Bcl-xl Axis. Front. Genet. 2020, 11, 536854. [Google Scholar] [CrossRef] [PubMed]
- Bakhdar, F.A.; Abdel Kawy, H.S.; Magadmi, R.M.; El-Kordy, E.A.; Alamri, A.S. Effect of histone deacetylase inhibitor (vorinostat) on new-onset diabetes induced by tacrolimus. J. Taibah Univ. Med. Sci. 2023, 18, 9–18. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, J.; Lin, R.; Huang, Y.; Wang, Z.; Xu, S.; Wang, L.; Chen, F.; Zhang, J.; Pan, K.; et al. Lactate drives epithelial-mesenchymal transition in diabetic kidney disease via the H3K14la/KLF5 pathway. Redox Biol. 2024, 75, 103246. [Google Scholar] [CrossRef]
- Zheng, T.; Gu, Y.P.; Wang, J.M.; Huang, T.T.; Gou, L.S.; Liu, Y.W. Lactate-triggered histone lactylation contributes to podocyte epithelial-mesenchymal transition in diabetic nephropathy in mice. Chem. Biol. Interact. 2025, 408, 111418. [Google Scholar] [CrossRef]
- Biessels, G.J.; Despa, F. Cognitive decline and dementia in diabetes mellitus: Mechanisms and clinical implications. Nat. Rev. Endocrinol. 2018, 14, 591–604. [Google Scholar] [CrossRef]
- Yang, Y.; Song, L.; Yu, L.; Zhang, J.; Zhang, B. H4K12 lactylation potentiates mitochondrial oxidative stress via the Foxo1 pathway in diabetes-induced cognitive impairment. J. Adv. Res. 2025. ahead of print. [Google Scholar] [CrossRef]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiełbowski, K.; Bakinowska, E.; Pawlik, A. Epigenetics Plays a Role in the Pathogenesis and Treatment of Diabetes. Genes 2025, 16, 769. https://doi.org/10.3390/genes16070769
Kiełbowski K, Bakinowska E, Pawlik A. Epigenetics Plays a Role in the Pathogenesis and Treatment of Diabetes. Genes. 2025; 16(7):769. https://doi.org/10.3390/genes16070769
Chicago/Turabian StyleKiełbowski, Kajetan, Estera Bakinowska, and Andrzej Pawlik. 2025. "Epigenetics Plays a Role in the Pathogenesis and Treatment of Diabetes" Genes 16, no. 7: 769. https://doi.org/10.3390/genes16070769
APA StyleKiełbowski, K., Bakinowska, E., & Pawlik, A. (2025). Epigenetics Plays a Role in the Pathogenesis and Treatment of Diabetes. Genes, 16(7), 769. https://doi.org/10.3390/genes16070769