
Characterizing Gene-Level Adaptations in the Gut Microbiome During Viral Infections: The Role of a Fucoidan-Rich Extract



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Human Safety Assessment
2.3. Study Design
2.4. Sample Size
2.5. Study Population
- Group A: 125 mg SLE-F, twice daily (n = 29; 32.2%).
- Group B: 500 mg SLE-F, twice daily (n = 30; 33.3%).
- Group C: Placebo, twice daily (n = 31; 34.4%).
2.6. Sample Collection
2.7. 16S rRNA Gene Metagenomic Sequencing
- Forward (IlluminaF): CCTACGGGNGGCWGCAG.
- Reverse (IlluminaR): GACTACHVGGGTATCTAATCC.
2.8. Bioinformatic Processing and Quality Control
2.9. Alpha Diversity Analysis
2.10. Random Forest Classifier Analysis
2.11. Recovery Score
2.12. Abundance and Biomarker Analysis
2.13. Statistical Analysis: LEfSe and Kruskal–Wallis Tests
2.13.1. LEfSe Analysis
2.13.2. Kruskal–Wallis H Test (Independent Analysis)
2.14. Statistical Analysis
3. Results
3.1. Epidemiological Context and Participant Infection Status
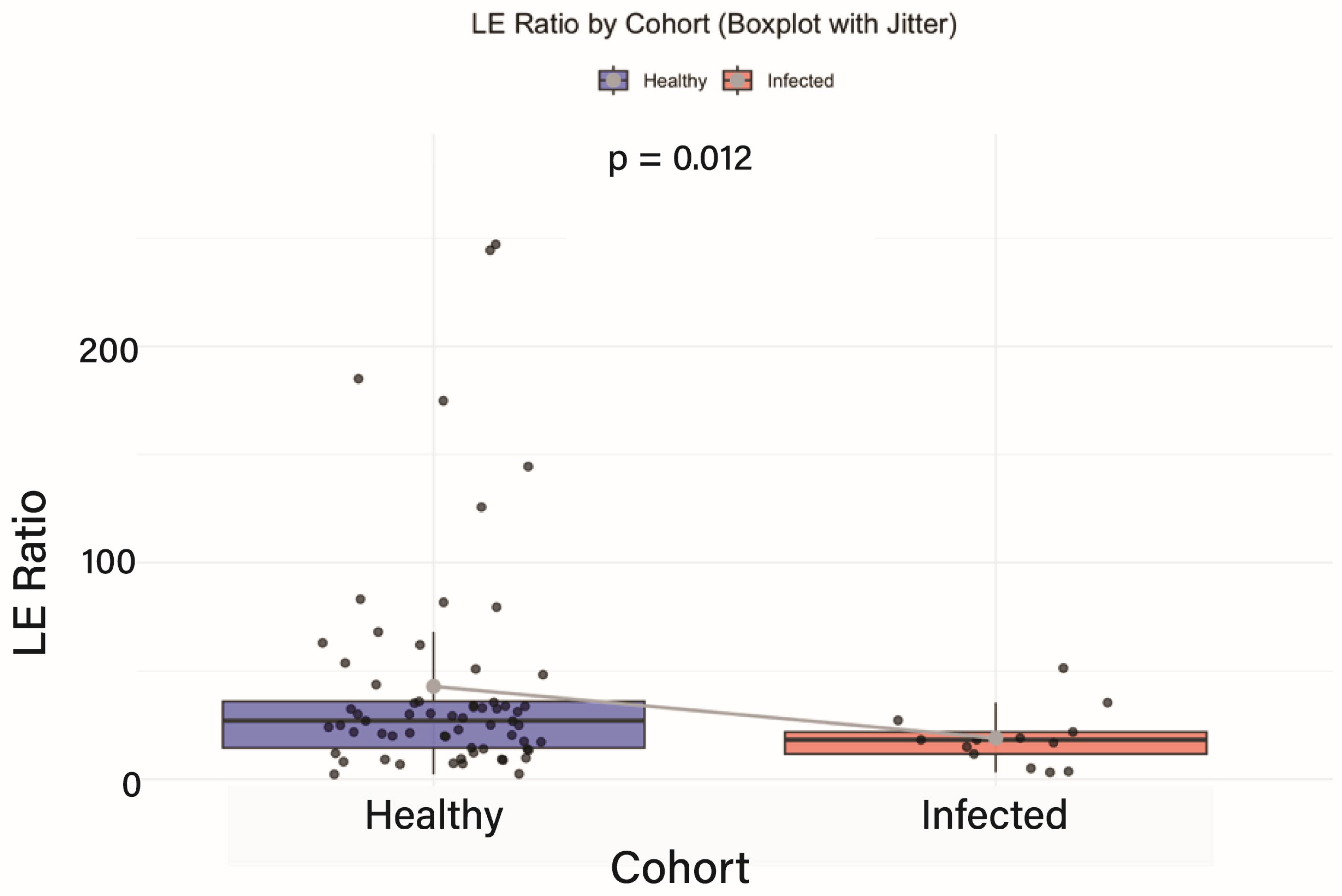
3.2. Baseline Comparison of Intestinal Inflammation
3.3. Functional Gene Signatures: Random Forest
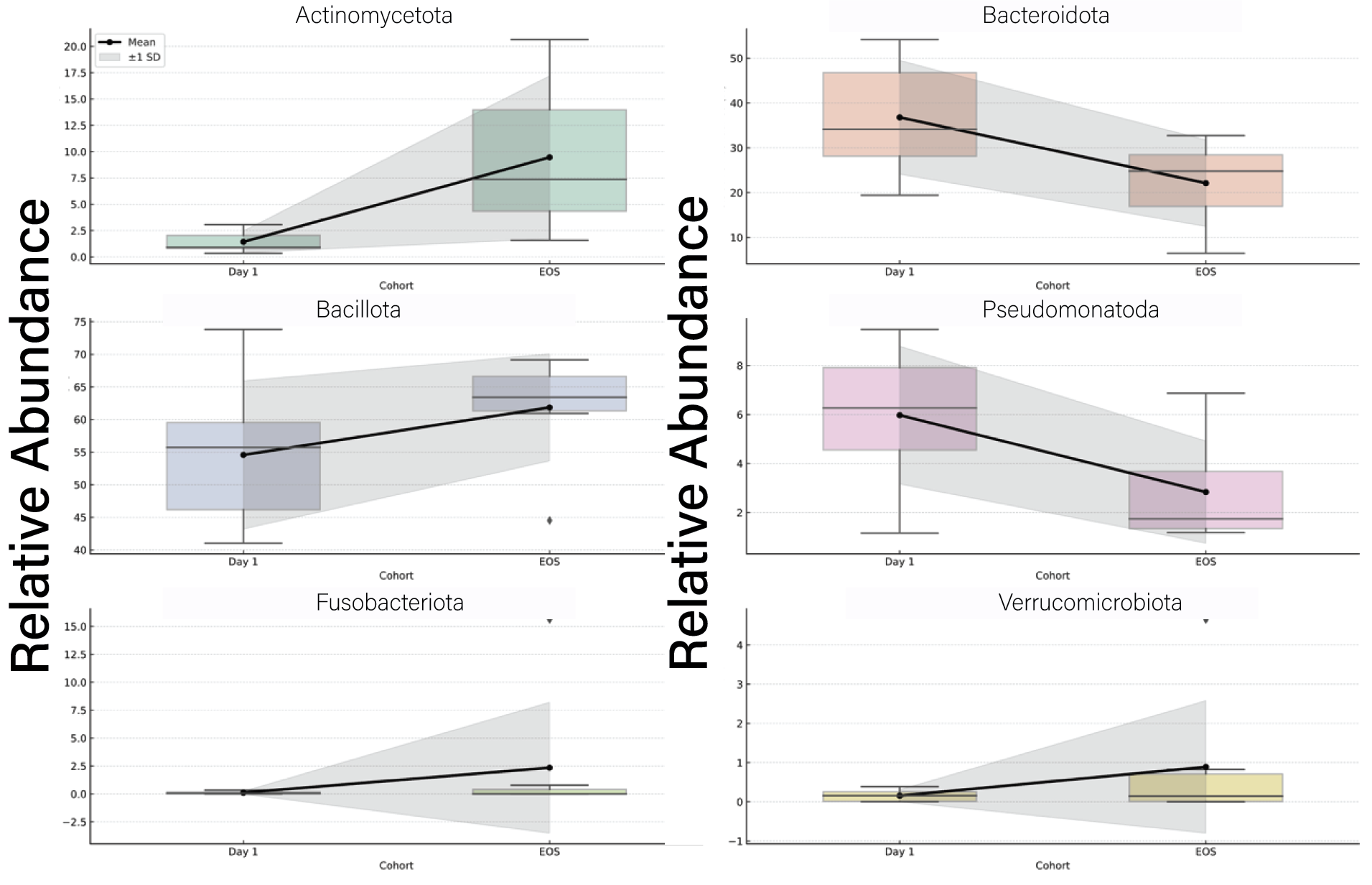
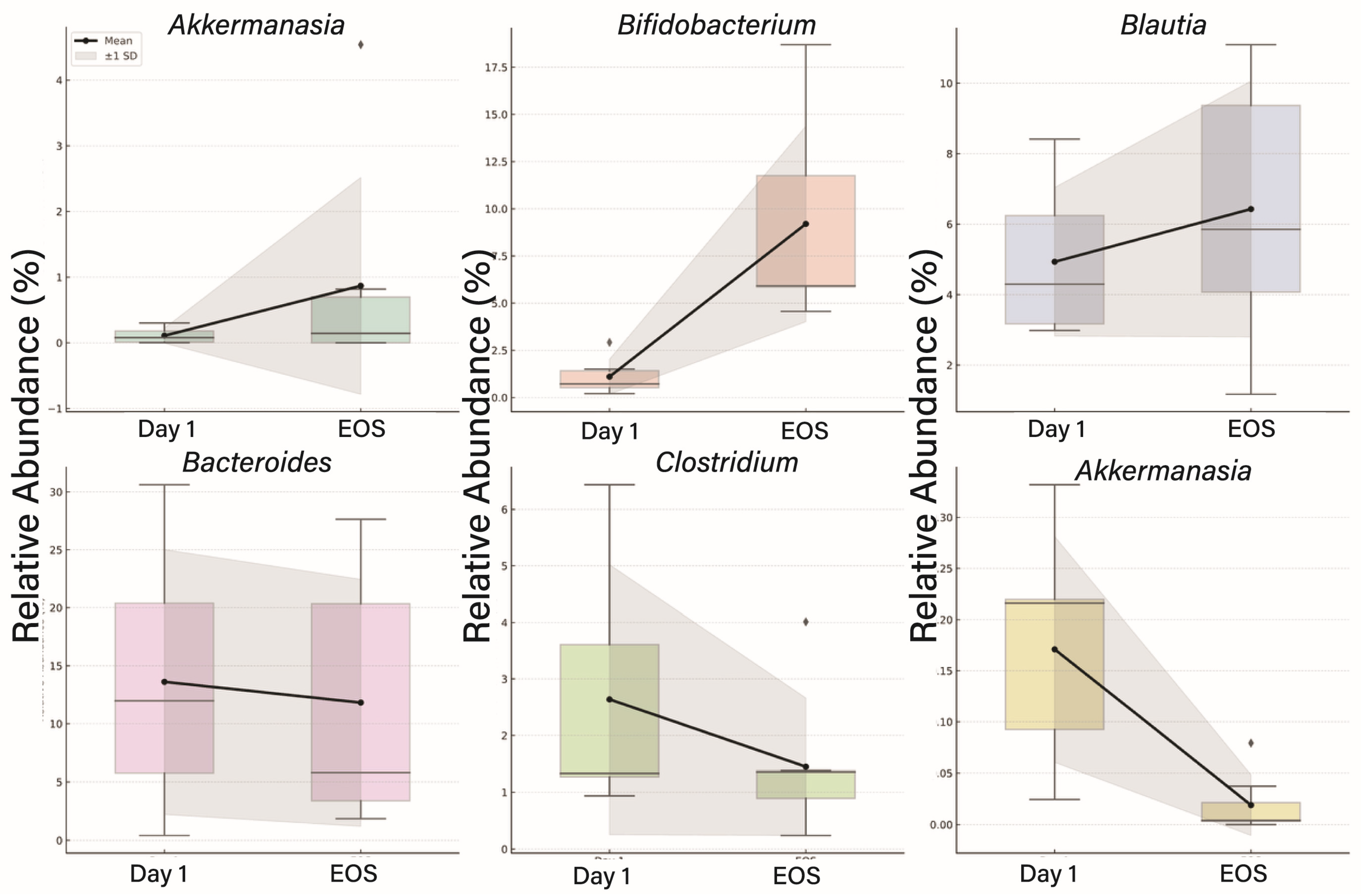
3.4. Taxonomic Shifts in the Gut Microbiome Following Viral Infection
3.5. Alpha Diversity Trends
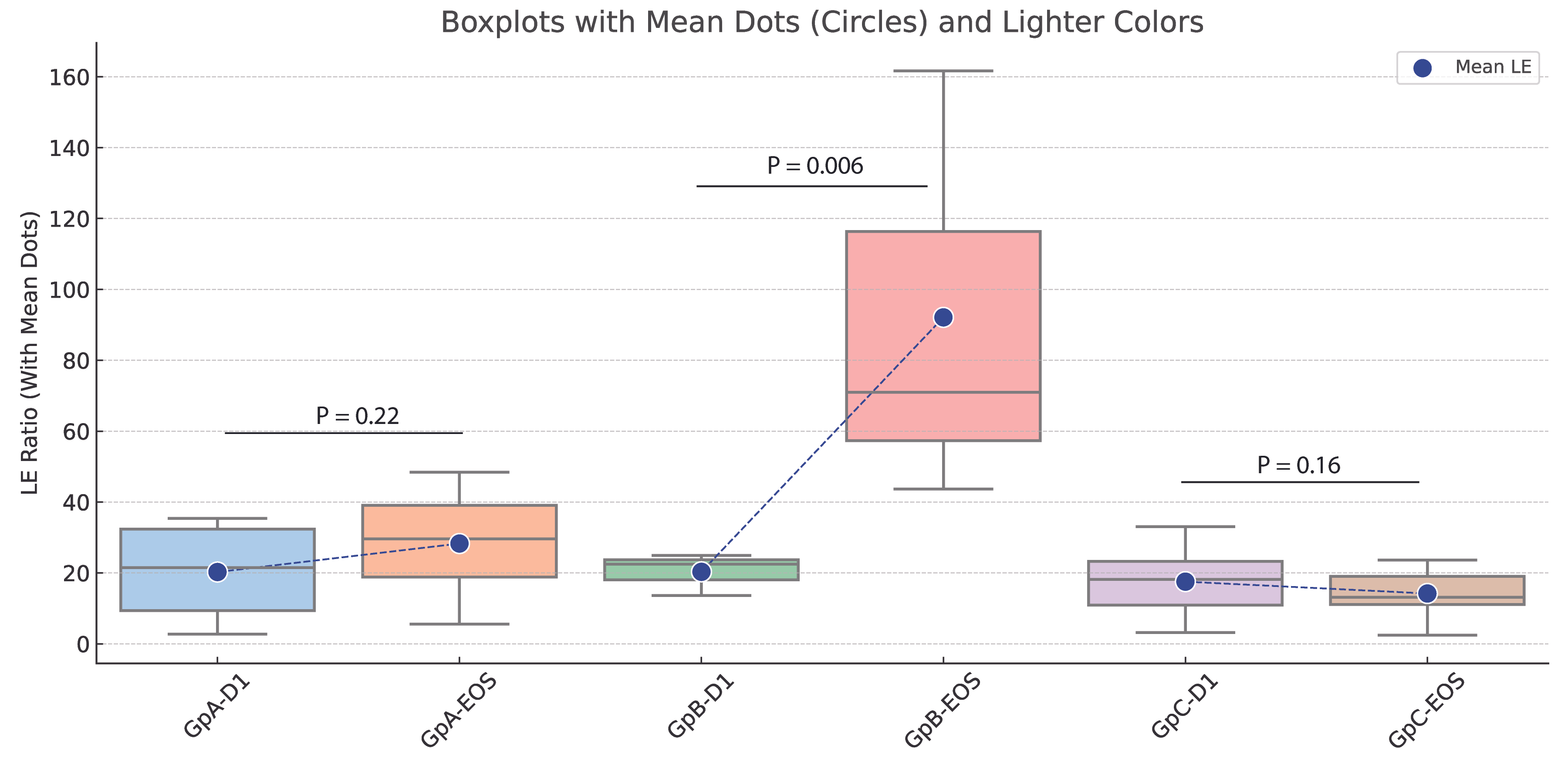
3.6. Impact of Fucoidan Treatment on LE Ratios and Functional Gene Profiles
4. Discussion
4.1. Baseline Comparison of Intestinal Inflammation
4.2. The Random Forest Model
4.3. Functional Gene Signatures Differentiate Fucoidan and Placebo Cohorts
4.4. Kruskal–Wallis Analysis: Suppression of Dysbiosis-Associated Functions in Fucoidan-Treated Participants
4.5. LEfSe Analysis: Promotion of Functional Resilience in the Fucoidan Group
Expanded Pathway-Level Insights from High-Dose Fucoidan EOS Samples
4.6. Functional Interpretation of Gene-Level Shifts in the Context of Taxonomy and Function
4.7. Integrative Insight and Therapeutic Implications
4.8. Diversity
4.9. Recovery Explanation
Microbial Activation Pathway
4.10. Limitations
- Subset Analysis: The observation regarding microbiome recovery post-viral infection was derived from a subset of participants who became infected during the trial. This subset was not pre-stratified or powered specifically to assess the effects of viral infection on microbiome dynamics, limiting the generalizability of the findings.
- Sample Size and Statistical Power: The number of participants infected with Dengue or Oropouche viruses within each treatment group was relatively small. This limited the statistical power for subgroup comparisons and may have contributed to the lack of significance in some outcomes, particularly in diversity measures and certain taxa-level analyses.
- Timing and Heterogeneity of Infection: Participants contracted viral infections at different time points during the study, which introduces heterogeneity in terms of exposure duration and immune response. Additionally, infections were not experimentally induced or uniformly documented via molecular diagnostics, which could influence the precision of infection status classification.
- Confounding by Other Variables: Although dietary and medication use exclusions were applied, other unmeasured factors—such as variations in baseline diet, host genetics, or environmental exposures—may have influenced microbiome composition and recovery trajectories.
- Short-Term Follow-Up: The study duration of 90 days may not have been sufficient to capture long-term microbiome stabilization or delayed effects of viral infection or SLE-F treatment. Longer follow-up would help clarify whether observed improvements persist over time.
- Taxonomic Resolution: While 16S rRNA sequencing offers valuable insights into bacterial composition, it lacks the resolution of metagenomic or metatranscriptomic approaches, limiting functional inference and the ability to capture strain-level variation.
- Lack of Virome and Mycobiome Profiling: The study focused exclusively on bacterial communities and did not assess viral or fungal components of the microbiome, which may also play critical roles in gut health and immune regulation during infection.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gomaa, E.Z. Human gut microbiota/microbiome in health and diseases: A review. Antonie Leeuwenhoek 2020, 113, 2019–2040. [Google Scholar] [CrossRef]
- Chancharoenthana, W.; Kamolratanakul, S.; Ariyanon, W.; Thanachartwet, V.; Phumratanaprapin, W.; Wilairatana, P.; Leelahavanichkul, A. Abnormal blood bacteriome, gut dysbiosis, and progression to severe dengue disease. Front. Cell. Infect. Microbiol. 2022, 12, 890817. [Google Scholar] [CrossRef] [PubMed]
- Perveen, N.; Muhammad, K.; Muzaffar, S.B.; Zaheer, T.; Munawar, N.; Gajic, B.; Sparagano, O.A.; Kishore, U.; Willingham, A.L. Host-pathogen interaction in arthropod vectors: Lessons from viral infections. Front. Immunol. 2023, 14, 1061899. [Google Scholar] [CrossRef] [PubMed]
- Pedreañez, A.; Carrero, Y.; Vargas, R.; Hernandez-Fonseca, J.P.; Hernandez-Fonseca, H.; Mosquera, J.A. Role of Gut Microbiota in Dengue. Rev. Med. Virol. 2024, 34, e2577. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Liang, H.; Li, M.; Chen, J.; Zhou, W.; Xia, D.; Ding, Q.; Yang, Y.; Zhang, Z.; Ran, C.; Zhou, Z. The intestinal microbiome and Cetobacterium somerae inhibit viral infection through TLR2-type I IFN signaling axis in zebrafish. Microbiome 2024, 12, 244. [Google Scholar] [CrossRef]
- Toledo, M.E.; Diaz, S.M.; Calderón, T.M.; Kreppel, K.; Van Damme, E.; Vanlerberghe, V. Preparedness for emerging epidemic threats: Detection of Oropouche circulation in Cuba. Lancet Infect. Dis. 2024, 24, e484. [Google Scholar] [CrossRef]
- Organización Panamericana de la Salud; Organización Mundial de la Salud. Actualización Epidemiológica: Oropouche en la Región de Las Américas, 6 de Marzo del 2024; OPS/OMS: Washington, DC, USA, 2024. [Google Scholar]
- Epidemiológica, P.d.B.H.V. Dengue. Balanço Dengue 2024, 5, 24. [Google Scholar]
- Fitton, J.H. Therapies from fucoidan; multifunctional marine polymers. Mar. Drugs 2011, 9, 1731–1760. [Google Scholar] [CrossRef]
- Li, B.; Lu, F.; Wei, X.; Zhao, R. Fucoidan: Structure and bioactivity. Molecules 2008, 13, 1671–1695. [Google Scholar] [CrossRef]
- Wijesekara, I.; Pangestuti, R.; Kim, S.-K. Biological activities and potential health benefits of sulfated polysaccharides derived from marine algae. Carbohydr. Polym. 2011, 84, 14–21. [Google Scholar] [CrossRef]
- Rioux, L.-E.; Turgeon, S.L.; Beaulieu, M. Structural characterization of laminaran and galactofucan extracted from the brown seaweed Saccharina longicruris. Phytochemistry 2010, 71, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- Fitton, J.H.; Stringer, D.N.; Park, A.Y.; Karpiniec, S.S. Therapies from fucoidan: New developments. Mar. Drugs 2019, 17, 571. [Google Scholar] [CrossRef] [PubMed]
- Makki, K.; Deehan, E.C.; Walter, J.; Bäckhed, F. The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe 2018, 23, 705–715. [Google Scholar] [CrossRef]
- Yin, H.; Li, R.; Liu, J.; Sun, Y.; Zhao, L.; Mou, J.; Yang, J. Fucosylated chondroitin sulfate from sea cucumber Stichopus chloronotus alleviate the intestinal barrier injury and oxidative stress damage in vitro and in vivo. Carbohydr. Polym. 2024, 328, 121722. [Google Scholar] [CrossRef]
- Hayashi, K.; Nakano, T.; Hashimoto, M.; Kanekiyo, K.; Hayashi, T. Defensive effects of a fucoidan from brown alga Undaria pinnatifida against herpes simplex virus infection. Int. Immunopharmacol. 2008, 8, 109–116. [Google Scholar] [CrossRef]
- Homer, B.; Barekatain, R.; Petrovski, K.R.; Plush, K.J.; Dwan, C.; D’Souza, D.N.; Verma, P.J.; Kirkwood, R.N.; Tucker, B.S. Preweaning Purified Fucoidan Drench: Effects on Growth, Immune Response, and Intestinal Morphology in Weaned Piglets. Animals 2024, 14, 1472. [Google Scholar] [CrossRef]
- Cano, R.; García, G. The Cuban Microbiome: Insights into Health, Disease, and the Role of Diet in Microbiome Recovery. An. Acad. Cienc. Cuba 2025, 15, 1–12. [Google Scholar]
- García, G.; Soto, J.; Netherland, M., Jr.; Hasan, N.A.; Buchaca, E.; Martínez, D.; Carlin, M.; de Jesus Cano, R. Evaluating the Effects of Sugar Shift® Symbiotic on Microbiome Composition and LPS Regulation: A Double-Blind, Placebo-Controlled Study. Microorganisms 2024, 12, 2525. [Google Scholar] [CrossRef]
- Vilagut, G.; Ferrer, M.; Rajmil, L.; Rebollo, P.; Permanyer-Miralda, G.; Quintana, J.M.; Santed, R.; Valderas, J.M.; Domingo-Salvany, A.; Alonso, J. El Cuestionario de Salud SF-36 español: Una década de experiencia y nuevos desarrollos. Gac. Sanit. 2005, 19, 135–150. [Google Scholar] [CrossRef]
- World Medical Association. World Medical Association Declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA 2013, 310, 2191–2194. [Google Scholar] [CrossRef] [PubMed]
- Schuck, L. Question Pro Survey Creation Software; Western Michigan University: Kalamazoo, MI, USA, 2012. [Google Scholar]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Nearing, J.T.; Douglas, G.M.; Comeau, A.M.; Langille, M.G. Denoising the Denoisers: An independent evaluation of microbiome sequence error-correction approaches. PeerJ 2018, 6, e5364. [Google Scholar] [CrossRef]
- Hall, M.; Beiko, R.G. 16S rRNA gene analysis with QIIME2. In Microbiome Analysis: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2018; pp. 113–129. [Google Scholar]
- Daniel, W.W. Kruskal–Wallis one-way analysis of variance by ranks. In Applied Nonparametric Statistics; PWS-Kent: Boston, MA, USA, 1990; pp. 226–234. [Google Scholar]
- Vázquez-Baeza, Y.; Pirrung, M.; Gonzalez, A.; Knight, R. EMPeror: A tool for visualizing high-throughput microbial community data. Gigascience 2013, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An ordination of upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Waldron, L.; Ballarini, A.; Narasimhan, V.; Jousson, O.; Huttenhower, C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 2012, 9, 811–814. [Google Scholar] [CrossRef] [PubMed]
- Rosner, B.; Glynn, R.J.; Lee, M.L.T. The Wilcoxon signed rank test for paired comparisons of clustered data. Biometrics 2006, 62, 185–192. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Benitez, A.J.; Alvarez, M.; Perez, L.; Gravier, R.; Serrano, S.; Hernandez, D.M.; Perez, M.M.; Gutierrez-Bugallo, G.; Martinez, Y.; Companioni, A.; et al. Oropouche Fever, Cuba, May 2024. Emerg. Infect. Dis. 2024, 30, 2155–2159. [Google Scholar] [CrossRef]
- World Health Organization. Disease Outbreak News; Oropouche Virus Disease in Cuba; Volume 521; World Health Organization: New York, NY, USA, 2024. [Google Scholar]
- Organización Panamericana de la Salud; Organización Mundial de la Salud. Actualización Epidemiológica: Dengue en la Región de las Américas 18 de Junio del 2024; PAHO/WHO: Washington, DC, USA, 2024. [Google Scholar]
- Organización Panamericana de la Salud; Organización Mundial de la Salud. Alerta Epidemiológica—SARS-CoV-2, Influenza y Otros Virus Respiratorios en la Región de las Américas—5 de Agosto del 2024; OPS/OMS: Washington, DC, USA, 2024. [Google Scholar]
- Krishnamurthy, H.K.; Pereira, M.; Bosco, J.; George, J.; Jayaraman, V.; Krishna, K.; Wang, T.; Bei, K.; Rajasekaran, J.J. Gut commensals and their metabolites in health and disease. Front. Microbiol. 2023, 14, 1244293. [Google Scholar] [CrossRef]
- Narayanan, S.A.; Jamison, D.A., Jr.; Guarnieri, J.W.; Zaksas, V.; Topper, M.; Koutnik, A.P.; Park, J.; Clark, K.B.; Enguita, F.J.; Leitão, A.L. A comprehensive SARS-CoV-2 and COVID-19 review, Part 2: Host extracellular to systemic effects of SARS-CoV-2 infection. Eur. J. Hum. Genet. 2024, 32, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Gasmi, A.; Mujawdiya, P.K.; Pivina, L.; Doşa, A.; Semenova, Y.; Benahmed, A.G.; Bjørklund, G. Relationship between gut microbiota, gut hyperpermeability and obesity. Curr. Med. Chem. 2021, 28, 827–839. [Google Scholar] [CrossRef]
- Shu, L.-Z.; Ding, Y.-D.; Xue, Q.-M.; Cai, W.; Deng, H. Direct and indirect effects of pathogenic bacteria on the integrity of intestinal barrier. Ther. Adv. Gastroenterol. 2023, 16, 17562848231176427. [Google Scholar] [CrossRef]
- Valiente, E.; Bouché, L.; Hitchen, P.; Faulds-Pain, A.; Songane, M.; Dawson, L.F.; Donahue, E.; Stabler, R.A.; Panico, M.; Morris, H.R. Role of glycosyltransferases modifying type B flagellin of emerging hypervirulent Clostridium difficile lineages and their impact on motility and biofilm formation. J. Biol. Chem. 2016, 291, 25450–25461. [Google Scholar] [CrossRef]
- Dai, X.; Zhu, M. Coupling of ribosome synthesis and translational capacity with cell growth. Trends Biochem. Sci. 2020, 45, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xie, J.; Feng, Z.; Ma, L.; Wu, W.; Guo, C.; He, J. Genomic insights into the cold adaptation and secondary metabolite potential of Pseudoalteromonas sp. WY3 from Antarctic krill. Front. Microbiol. 2024, 15, 1459716. [Google Scholar] [CrossRef]
- Becerra, J.E.; Yebra, M.J.; Monedero, V. An L-fucose operon in the probiotic Lactobacillus rhamnosus GG is involved in adaptation to gastrointestinal conditions. Appl. Environ. Microbiol. 2015, 81, 3880–3888. [Google Scholar] [CrossRef] [PubMed]
- Zaplana, T.; Miele, S.; Tolonen, A.C. Lachnospiraceae are emerging industrial biocatalysts and biotherapeutics. Front. Bioeng. Biotechnol. 2024, 11, 1324396. [Google Scholar] [CrossRef]
- Li, N.; Ma, W.-T.; Pang, M.; Fan, Q.-L.; Hua, J.-L. The commensal microbiota and viral infection: A comprehensive review. Front. Immunol. 2019, 10, 1551. [Google Scholar] [CrossRef]
- Wirusanti, N.I.; Baldridge, M.T.; Harris, V.C. Microbiota regulation of viral infections through interferon signaling. Trends Microbiol. 2022, 30, 778–792. [Google Scholar] [CrossRef]
- Campbell, D.E.; Li, Y.; Ingle, H.; Baldridge, M.T. Impact of the microbiota on viral infections. Annu. Rev. Virol. 2023, 10, 371–395. [Google Scholar] [CrossRef]
- Yang, M.; Yang, Y.; He, Q.; Zhu, P.; Liu, M.; Xu, J.; Zhao, M. Intestinal Microbiota—A Promising Target for Antiviral Therapy? Front. Immunol. 2021, 12, 676232. [Google Scholar] [CrossRef] [PubMed]
- Guédon, G.; Charron-Bourgoin, F.; Lacroix, T.; Hamadouche, T.; Soler, N.; Douzi, B.; Chiapello, H.; Leblond-Bourget, N. Massive acquisition of conjugative and mobilizable integrated elements fuels Faecalibacterium plasticity and hints at their adaptation to the gut. Sci. Rep. 2025, 15, 17013. [Google Scholar] [CrossRef]
- Robinson, C.R.; Dolezal, A.G.; Liachko, I.; Newton, I.L. Mobile genetic elements exhibit associated patterns of host range variation and sequence diversity within the gut microbiome of the European Honey bee. bioRxiv 2025. [Google Scholar] [CrossRef]
- Almeida, A.; Nayfach, S.; Boland, M.; Strozzi, F.; Beracochea, M.; Shi, Z.J.; Pollard, K.S.; Sakharova, E.; Parks, D.H.; Hugenholtz, P. A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol. 2021, 39, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Marcos-Zambrano, L.J.; Karaduzovic-Hadziabdic, K.; Loncar Turukalo, T.; Przymus, P.; Trajkovik, V.; Aasmets, O.; Berland, M.; Gruca, A.; Hasic, J.; Hron, K. Applications of machine learning in human microbiome studies: A review on feature selection, biomarker identification, disease prediction and treatment. Front. Microbiol. 2021, 12, 634511. [Google Scholar] [CrossRef] [PubMed]
- Arita, K.; Nakano, Y.; Kasai, S.; Yoshizawa, K.; Miyazaki, S. Computational Identification of Specific Genes and Those Functions on the Gut Microbiota. Preprints 2025, 2025011007. [Google Scholar] [CrossRef]
- Moreira de Gouveia, M.I.; Bernalier-Donadille, A.; Jubelin, G. Enterobacteriaceae in the human gut: Dynamics and ecological roles in health and disease. Biology 2024, 13, 142. [Google Scholar] [CrossRef]
- Winter, S.E.; Bäumler, A.J. Gut dysbiosis: Ecological causes and causative effects on human disease. Proc. Natl. Acad. Sci. USA 2023, 120, e2316579120. [Google Scholar] [CrossRef]
- Baldelli, V.; Scaldaferri, F.; Putignani, L.; Del Chierico, F. The role of Enterobacteriaceae in gut microbiota dysbiosis in inflammatory bowel diseases. Microorganisms 2021, 9, 697. [Google Scholar] [CrossRef]
- Maciel-Fiuza, M.F.; Muller, G.C.; Campos, D.M.S.; do Socorro Silva Costa, P.; Peruzzo, J.; Bonamigo, R.R.; Veit, T.; Vianna, F.S.L. Role of gut microbiota in infectious and inflammatory diseases. Front. Microbiol. 2023, 14, 1098386. [Google Scholar] [CrossRef]
- Ferrocino, I.; Rantsiou, K.; McClure, R.; Kostic, T.; de Souza, R.S.C.; Lange, L.; FitzGerald, J.; Kriaa, A.; Cotter, P.; Maguin, E. The need for an integrated multi-OMICs approach in microbiome science in the food system. Compr. Rev. Food Sci. Food Saf. 2023, 22, 1082–1103. [Google Scholar] [CrossRef]
- Zhang, Y.; Thomas, J.P.; Korcsmaros, T.; Gul, L. Integrating multi-omics to unravel host-microbiome interactions in inflammatory bowel disease. Cell Rep. Med. 2024, 5, 101738. [Google Scholar] [CrossRef]
- Farah, A.; Paul, P.; Khan, A.S.; Sarkar, A.; Laws, S.A.; Chaari, A. Targeting gut microbiota dysbiosis in inflammatory bowel disease: A systematic review of current evidence. Front. Med. 2025, 12, 1435030. [Google Scholar] [CrossRef]
- Xie, H.; Yu, S.; Tang, M.-Y.; Xun, Y.; Shen, Q.; Wu, G. Gut Microbiota Dysbiosis in Inflammatory Bowel Disease: Interaction with Intestinal Barriers and Microbiota-targeted Treatment Options. Front. Cell. Infect. Microbiol. 2025, 15, 1608025. [Google Scholar]
- Yan, Y.; Cao, M.; Ma, J.; Suo, J.; Bai, X.; Ge, W.; Lü, X.; Zhang, Q.; Chen, J.; Cui, S. Mechanisms of thermal, acid, desiccation and osmotic tolerance of Cronobacter spp. Crit. Rev. Food Sci. Nutr. 2024, 1–23. [Google Scholar] [CrossRef]
- Wizenty, J.; Sigal, M. Helicobacter pylori, microbiota and gastric cancer—Principles of microorganism-driven carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2025, 22, 296–313. [Google Scholar] [CrossRef]
- Willett, J.L.; Dunny, G.M. Insights into ecology, pathogenesis, and biofilm formation of Enterococcus faecalis from functional genomics. Microbiol. Mol. Biol. Rev. 2025, 89, e00081-00023. [Google Scholar] [CrossRef]
- Silva-Santana, G.; Sales, F.L.S.; Aguiar, A.R.; Brandão, M.L.L. Pharmaceutical Contamination by Biofilms Formed of the Burkholderia cepacia Complex: Public Health Risks. Processes 2025, 13, 1270. [Google Scholar] [CrossRef]
- Wimmer, B.C.; Dwan, C.; De Medts, J.; Duysburgh, C.; Rotsaert, C.; Marzorati, M. Undaria pinnatifida Fucoidan Enhances Gut Microbiome, Butyrate Production, and Exerts Anti-Inflammatory Effects in an In Vitro Short-Term SHIME® Coupled to a Caco-2/THP-1 Co-Culture Model. Mar. Drugs 2025, 23, 242. [Google Scholar] [CrossRef]
- Wang, H.; Wei, W.; Liu, F.; Wang, M.; Zhang, Y.; Du, S. Effects of fucoidan and synbiotics supplementation during bismuth quadruple therapy of Helicobacter pylori infection on gut microbial homeostasis: An open-label, randomized clinical trial. Front. Nutr. 2024, 11, 1407736. [Google Scholar] [CrossRef] [PubMed]
- Hermes, G.D.; Rasmussen, C.; Wellejus, A. Variation in the conservation of species-specific gene sets for HMO degradation and its effects on HMO utilization in bifidobacteria. Nutrients 2024, 16, 1893. [Google Scholar] [CrossRef]
- CB, L.; Ghosh, T.S. Microbiome Surfaceome and Secretome Atlas: Building a Dictionary of Putative Surface and Secreted Proteins on the Human Microbiome; IIIT-Delhi: New Delhi, India, 2024. [Google Scholar]
- Bhagwat, A.; Haldar, T.; Kanojiya, P.; Saroj, S.D. Bacterial metabolism in the host and its association with virulence. Virulence 2025, 16, 2459336. [Google Scholar] [CrossRef]
- Basu, M. Investigating the Host-Associated Microbiome in Biomedicine: Insights from Experimental Models, Host Genotype Effects, and Disease Implications. Ph.D. Thesis, Kiel University, Kiel, Germany, 2024. [Google Scholar]
- Méndez, V.; Sepúlveda, M.; Izquierdo-Fiallo, K.; Macaya, C.C.; Esparza, T.; Báez-Matus, X.; Durán, R.E.; Levicán, G.; Seeger, M. Surfing in the storm: How Paraburkholderia xenovorans thrives under stress during biodegradation of toxic aromatic compounds and other stressors. FEMS Microbiol. Rev. 2025, 49, fuaf021. [Google Scholar] [CrossRef]
- Dang, H.J. Exploring Kinetic Controlled Protein Solubility Under Physiologically Relevant Conditions; The University of Wisconsin-Madison: Madison, WI, USA, 2024. [Google Scholar]
- Fan, S.; Zhang, Z.; Nie, Q.; Ackah, M.; Nie, S. Rethinking the classification of non-digestible carbohydrates: Perspectives from the gut microbiome. Compr. Rev. Food Sci. Food Saf. 2024, 23, e70046. [Google Scholar] [CrossRef]
- Xiao, M.; Zhang, C.; Duan, H.; Narbad, A.; Zhao, J.; Chen, W.; Zhai, Q.; Yu, L.; Tian, F. Cross-feeding of bifidobacteria promotes intestinal homeostasis: A lifelong perspective on the host health. npj Biofilms Microbiomes 2024, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Guzior, D.V.; Martin, C.; Neugebauer, K.A.; Rzepka, M.M.; Lumeng, J.C.; Quinn, R.A.; de Los Campos, G. Longitudinal analyses of infants’ microbiome and metabolome reveal microbes and metabolites with seemingly coordinated dynamics. Commun. Biol. 2024, 7, 1506. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.K.; Chattopadhyay, D. Aging and DNA Damage: Investigating the Microbiome’s Stealthy Impact—A Perspective. Explor. Res. Hypothesis Med. 2025, 10, 106–121. [Google Scholar] [CrossRef]
- Turner, B.R.; Jenkinson, P.I.; Huttman, M.; Mullish, B.H. Inflammation, oxidative stress and gut microbiome perturbation: A narrative review of mechanisms and treatment of the alcohol hangover. Alcohol Clin. Exp. Res. 2024, 48, 1451–1465. [Google Scholar] [CrossRef]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.M.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef]
- Koropatkin, N.M.; Cameron, E.A.; Martens, E.C. How glycan metabolism shapes the human gut microbiota. Nat. Rev. Microbiol. 2012, 10, 323–335. [Google Scholar] [CrossRef]
- Schneewind, O.; Missiakas, D. Sec-secretion and sortase-mediated anchoring of proteins in Gram-positive bacteria. Biochim. Biophys. Acta 2014, 1843, 1687–1697. [Google Scholar] [CrossRef]
- Li, S.; Qian, Q.; Xie, Y.; Wu, Z.; Yang, H.; Yin, Y.; Cui, Y.; Li, X. Ameliorated Effects of Fucoidan on Dextran Sulfate Sodium-Induced Ulcerative Colitis and Accompanying Anxiety and Depressive Behaviors in Aged C57BL/6 Mice. J. Agric. Food Chem. 2024, 72, 14199–14215. [Google Scholar] [CrossRef]
- Macchione, I.; Lopetuso, L.R.; Ianiro, G.; Napoli, M.; Gibiino, G.; Rizzatti, G.; Petito, V.; Gasbarrini, A.; Scaldaferri, F. Akkermansia muciniphila: Key player in metabolic and gastrointestinal disorders. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8075–8083. [Google Scholar]
- Gavzy, S.J.; Kensiski, A.; Lee, Z.L.; Mongodin, E.F.; Ma, B.; Bromberg, J.S. Bifidobacterium mechanisms of immune modulation and tolerance. Gut Microbes 2023, 15, 2291164. [Google Scholar] [CrossRef] [PubMed]
- Sarita, B.; Samadhan, D.; Hassan, M.Z.; Kovaleva, E.G. A comprehensive review of probiotics and human health-current prospective and applications. Front. Microbiol. 2025, 15, 1487641. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, H.; Wu, P.; Yang, S.; Xue, W.; Xu, B.; Zhang, S.; Tang, B.; Xu, D. Akkermansia muciniphila: A promising probiotic against inflammation and metabolic disorders. Virulence 2024, 15, 2375555. [Google Scholar] [CrossRef]
- Ross, F.C.; Patangia, D.; Grimaud, G.; Lavelle, A.; Dempsey, E.M.; Ross, R.P.; Stanton, C. The interplay between diet and the gut microbiome: Implications for health and disease. Nat. Rev. Microbiol. 2024, 22, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Blattman, S.B.; Takahashi, M.; Mandayam, N.; Jiang, W.; Oikonomou, P.; Tavazoie, S.F.; Tavazoie, S. Conserved genetic basis for microbial colonization of the gut. Cell 2025, 188, 2505–2520.e22. [Google Scholar] [CrossRef]
- Lu, J.; Wei, W.; Zheng, D. Fusobacterium nucleatum in Colorectal Cancer: Ally Mechanism and Targeted Therapy Strategies. Research 2025, 8, 0640. [Google Scholar] [CrossRef]
- Shenoy, S.; Jena, A.; Levinson, C.; Sharma, V.; Deepak, P.; Aswani-Omprakash, T.; Sebastian, S.; Colombel, J.-F.; Agrawal, M. Inflammatory bowel disease in south Asia: A scoping review. Lancet Gastroenterol. Hepatol. 2025, 10, 259–274. [Google Scholar] [CrossRef]
- Zheng, W.; Tang, S.; Ren, X.; Song, S.; Ai, C. Fucoidan alleviated colitis aggravated by fiber deficiency through protecting the gut barrier, suppressing the MAPK/NF-κB pathway, and modulating gut microbiota and metabolites. Front. Nutr. 2025, 11, 1462584. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, H.; Xu, L.; Zhang, X.; Chen, W.; Wang, C. Therapeutic Potential of Fucoidan in Alleviating Histamine-Induced Liver Injury: Insights from Mice Studies. Foods 2024, 13, 1523. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, M.; Li, Z.; Zhao, D.; Hou, Y.; Wu, T. Fucoidan Alleviates Porcine Epidemic Diarrhea Virus-Induced Intestinal Damage in Piglets by Enhancing Antioxidant Capacity and Modulating Arginine Metabolism. Animals 2025, 15, 1001. [Google Scholar] [CrossRef]
- Khalili, L.; Park, G.; Nagpal, R.; Salazar, G. The role of Akkermansia muciniphila on improving gut and metabolic health modulation: A meta-analysis of preclinical mouse model studies. Microorganisms 2024, 12, 1627. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Qian, Q.; Yang, H.; Wu, Z.; Xie, Y.; Yin, Y.; Cui, Y.; Li, X. Fucoidan alleviated dextran sulfate sodium–induced ulcerative colitis with improved intestinal barrier, reshaped gut microbiota composition, and promoted autophagy in male C57BL/6 mice. Nutr. Res. 2024, 122, 1–18. [Google Scholar] [CrossRef]
- Hou, K.; Wu, Z.-X.; Chen, X.-Y.; Wang, J.-Q.; Zhang, D.; Xiao, C.; Zhu, D.; Koya, J.B.; Wei, L.; Li, J. Microbiota in health and diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Kumar, S.; Mukherjee, R.; Gaur, P.; Leal, É.; Lyu, X.; Ahmad, S.; Puri, P.; Chang, C.-M.; Raj, V.S.; Pandey, R.P. Unveiling roles of beneficial gut bacteria and optimal diets for health. Front. Microbiol. 2025, 16, 1527755. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Xiong, D.; Shi, J.; Long, M.; Chen, Z. The interaction between viruses and intestinal microbiota: A review. Curr. Microbiol. 2021, 78, 3597–3608. [Google Scholar] [CrossRef] [PubMed]
- Altomare, A.; Giovanetti, M.; Baldaro, F.; Ciccozzi, M.; Cicala, M.; Guarino, M.P.L. The prevention of viral infections: The role of intestinal microbiota and nutritional factors. Nutrients 2024, 16, 2445. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, D.; Chen, R.; Zhang, Y.; Wang, C.; Qian, G. Recent insights and advances in gut microbiota’s influence on host antiviral immunity. Front. Microbiol. 2025, 16, 1536778. [Google Scholar] [CrossRef]
- Xu, L.; Yang, C.S.; Liu, Y.; Zhang, X. Effective regulation of gut microbiota with probiotics and prebiotics may prevent or alleviate COVID-19 through the gut-lung axis. Front. Pharmacol. 2022, 13, 895193. [Google Scholar] [CrossRef]
- Akhtar, A.A.; Turner, D.P. The role of bacterial ATP-binding cassette (ABC) transporters in pathogenesis and virulence: Therapeutic and vaccine potential. Microb. Pathog. 2022, 171, 105734. [Google Scholar] [CrossRef]
- Locher, K.P. Structure and mechanism of ABC transporters. Curr. Opin. Struct. Biol. 2004, 14, 426–431. [Google Scholar] [CrossRef]
- Maresso, A.W.; Maresso, A.W. The Acquisition and Consumption of Host Nutrients. In Bacterial Virulence: A Conceptual Primer; Springer: Berlin/Heidelberg, Germany, 2019; pp. 131–144. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef]
- Safarchi, A.; Al-Qadami, G.; Tran, C.D.; Conlon, M. Understanding dysbiosis and resilience in the human gut microbiome: Biomarkers, interventions, and challenges. Front. Microbiol. 2025, 16, 1559521. [Google Scholar] [CrossRef] [PubMed]
- Zechner, E.L.; Lang, S.; Schildbach, J.F. Assembly and mechanisms of bacterial type IV secretion machines. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 1073–1087. [Google Scholar] [CrossRef]
- Sonnenburg, E.D.; Sonnenburg, J.L. The ancestral and industrialized gut microbiota and implications for human health. Nat. Rev. Microbiol. 2019, 17, 383–390. [Google Scholar] [CrossRef]
- Ze, X.; Duncan, S.H.; Louis, P.; Flint, H.J. Ruminococcus bromii is a keystone species for the degradation of resistant starch in the human colon. ISME J. 2012, 6, 1535–1543. [Google Scholar] [CrossRef]
- Javanshir, N.; Hosseini, G.N.G.; Sadeghi, M.; Esmaeili, R.; Satarikia, F.; Ahmadian, G.; Allahyari, N. Evaluation of the Function of Probiotics, Emphasizing the Role of their Binding to the Intestinal Epithelium in the Stability and their Effects on the Immune System. Biol. Proced. Online 2021, 23, 23. [Google Scholar] [CrossRef]
- Tavares, N.C.; Nascimento, C.S.; de Oliveira, J.G.; Calzavara-Silva, C.E. Exploring Host Factors of the Human Metabolism as Promising Targets for Dengue Treatment. In Viral Infectious Diseases Annual Volume 2024; IntechOpen: London, UK, 2024. [Google Scholar]
- Romani, F.E.; Luvira, V.; Chancharoenthana, W.; Albanese, M.; Maddaloni, L.; Branda, F.; D’Amelio, S.; Gabrielli, S.; Scagnolari, C.; Mastroianni, C.M. Human microbiota in dengue infection: A narrative review. Microb. Pathog. 2025, 205, 107643. [Google Scholar] [CrossRef]
- Jackson, J.J.; Heyer, S.; Bell, G. Sortase-encoding genes, srtA and srtC, mediate Enterococcus faecalis OG1RF persistence in the Helicoverpa zea gastrointestinal tract. Front. Microbiol. 2024, 15, 1322303. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | p-Value | ||||
|---|---|---|---|---|---|
| A | B | C | (χ2) | ||
| 29 | 30 | 31 | |||
| Viral infection during the study | No | 15 (51.7%) | 14 (46.7%) | 14 (45.2%) | 0.869 |
| yes | 14 (48.3%) | 16 (53.3%) | 17 (52.2%) | ||
| ↓ | ↓ | ↓ | ↓ | ||
| Before/Beginning | 4 (28.6%) | 6 (37.5%) | 10 (58.8%) | 0.207 | |
| During | 10 (71.4%) | 10 (62.5%) | 7 (41.2%) | ||
| Ortholog | Definition | Importance Score | SLE-F EOS | Placebo EOS | Enriched Group |
|---|---|---|---|---|---|
| K01186 | Sialidase | 0.0588 | 0.0560 | 0.0615 | Placebo |
| K10118 | raffinose/stachyose/melibiose transport system permease protein | 0.0588 | 0.0516 | 0.0455 | SLE-F |
| K08191 | MFS transporter, ACS family, hexuronate | 0.0588 | 0.0392 | 0.0156 | SLE-F |
| K06921 | uncharacterized protein | 0.0588 | 0.1570 | 0.0734 | SLE-F |
| K16785 | energy-coupling factor transport system permease protein | 0.0471 | 0.0535 | 0.0696 | Placebo |
| K19353 | heptose-I-phosphate ethanolaminephosphotransferase | 0.0431 | 0.0208 | 0.0076 | SLE-F |
| K21903 | ArsR family transcriptional regulator, lead/cadmium/zinc/bismuth-responsive transcriptional repressor | 0.0353 | 0.0279 | 0.0414 | Placebo |
| K12988 | alpha-1,3-rhamnosyltransferase | 0.0353 | 0.0378 | 0.0177 | SLE-F |
| K01462 | peptide deformylase | 0.0353 | 0.0756 | 0.0732 | SLE-F |
| K01607 | carboxymuconolactone | 0.0353 | 0.0452 | 0.0687 | Placebo |
| A. Kruskal–Wallis | |||||
| Ortholog | Definition | Fold Change SLE-F | Fold Change Placebo | FDR Adjusted p-Value | Cohort Enriched |
| K09885 | Aquaporin rerated protein, other eukaryotes | 0.0180 | 1.0000 | 0.0002 | Placebo |
| K03124 | Transcription initiation factor TFIIB | 0.0088 | 1.0000 | 0.0002 | Placebo |
| K15965 | glycosyltransferase | 0.0093 | 1.0000 | 0.0002 | Placebo |
| K05083 | Receptor tyrosine-protein kinase erbB-2 | 0.0180 | 0.1250 | 0.0028 | Placebo |
| K03334 | L-amino-acid oxidase | 0.0076 | 0.1272 | 0.0028 | Placebo |
| K21148 | [CysO sulfur-carrier protein]-thiocarboxylate-dependent cysteine synthase | 0.0053 | 0.1272 | 0.0028 | Placebo |
| K15761 | Toluene monooxygenase system protein B | 0.0138 | 0.1250 | 0.0028 | Placebo |
| K03773 | Protein-folding Isomerase | 0.0048 | 0.1272 | 0.0028 | Placebo |
| K02429 | L-fucose permease | 0.0066 | 0.1272 | 0.0028 | Placebo |
| K07190 | Phosphorylase kinase alpha/beta subunit | 0.0059 | 0.0673 | 0.0031 | Placebo |
| B. LEfSe (Linear Discriminant Analysis Effect Size) | |||||
| Ortholog | Definition | Fold Change SLE-F | Fold Change Placebo | LDA Effect Size | Cohort Enriched |
| K07284 | Sortase A | 1.2755 | 0.6212 | 2.6276 | SLE-F |
| K02315 | DNA replication protein DnaC | 1.5720 | 1.6683 | 2.4694 | Placebo |
| K03773 | FKBP-type peptidyl-prolyl cis-trans isomerase FklB | 1.0909 | 0.5163 | 2.4286 | SLE-F |
| K02003, K02004, K06147 | ABC transport system ATP-binding protein | 1.3726 | 1.1534 | 2.4191 | SLE-F |
| K02429 | MFS transporter, FHS family, L-fucose permease | 1.0626 | 0.5566 | 2.3667 | SLE-F |
| K02030 | Polar amino acid transport system substrate-binding protein | 1.2128 | 1.3541 | 2.2981 | Placebo |
| K03286 | OmpA-OmpF porin, OOP family | 1.0185 | 0.5629 | 2.2478 | SLE-F |
| K10117,K10118, K10119 | Raffinose/stachyose/melibiose transport system substrate-binding protein | 1.6093 | 1.4149 | 2.2456 | SLE-F |
| K03657 | Stress response/DNA helicase I/ATP-dependent DNA helicase PcrA | 1.2597 | 1.3317 | 2.1916 | SLE-F |
| K16785 | Energy-coupling factor transport system permease protein | 1.3868 | 1.8712 | 2.1724 | Placebo |
| KO1214 | Isoamylase | 1.7511 | 1.1182 | 2.6544 | SLE-F |
| KO0688 | Glycogen phosphorylase | 1.3336 | 1.1124 | 2.4281 | SLE-F |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García, G.; Soto, J.; Valenzuela, C.; Cano, R.D.J. Characterizing Gene-Level Adaptations in the Gut Microbiome During Viral Infections: The Role of a Fucoidan-Rich Extract. Genes 2025, 16, 740. https://doi.org/10.3390/genes16070740
García G, Soto J, Valenzuela C, Cano RDJ. Characterizing Gene-Level Adaptations in the Gut Microbiome During Viral Infections: The Role of a Fucoidan-Rich Extract. Genes. 2025; 16(7):740. https://doi.org/10.3390/genes16070740
Chicago/Turabian StyleGarcía, Gissel, Josanne Soto, Carmen Valenzuela, and Raul De Jesús Cano. 2025. "Characterizing Gene-Level Adaptations in the Gut Microbiome During Viral Infections: The Role of a Fucoidan-Rich Extract" Genes 16, no. 7: 740. https://doi.org/10.3390/genes16070740
APA StyleGarcía, G., Soto, J., Valenzuela, C., & Cano, R. D. J. (2025). Characterizing Gene-Level Adaptations in the Gut Microbiome During Viral Infections: The Role of a Fucoidan-Rich Extract. Genes, 16(7), 740. https://doi.org/10.3390/genes16070740