
Radiogenomics of Stereotactic Radiotherapy: Genetic Mechanisms Underlying Radiosensitivity, Resistance, and Immune Response

 , ,
, , 
Abstract
1. Introduction
2. Radiobiological Mechanisms of Stereotactic Body Radiation Therapy
2.1. Radiobiological Rationale of SBRT and the LQ Model
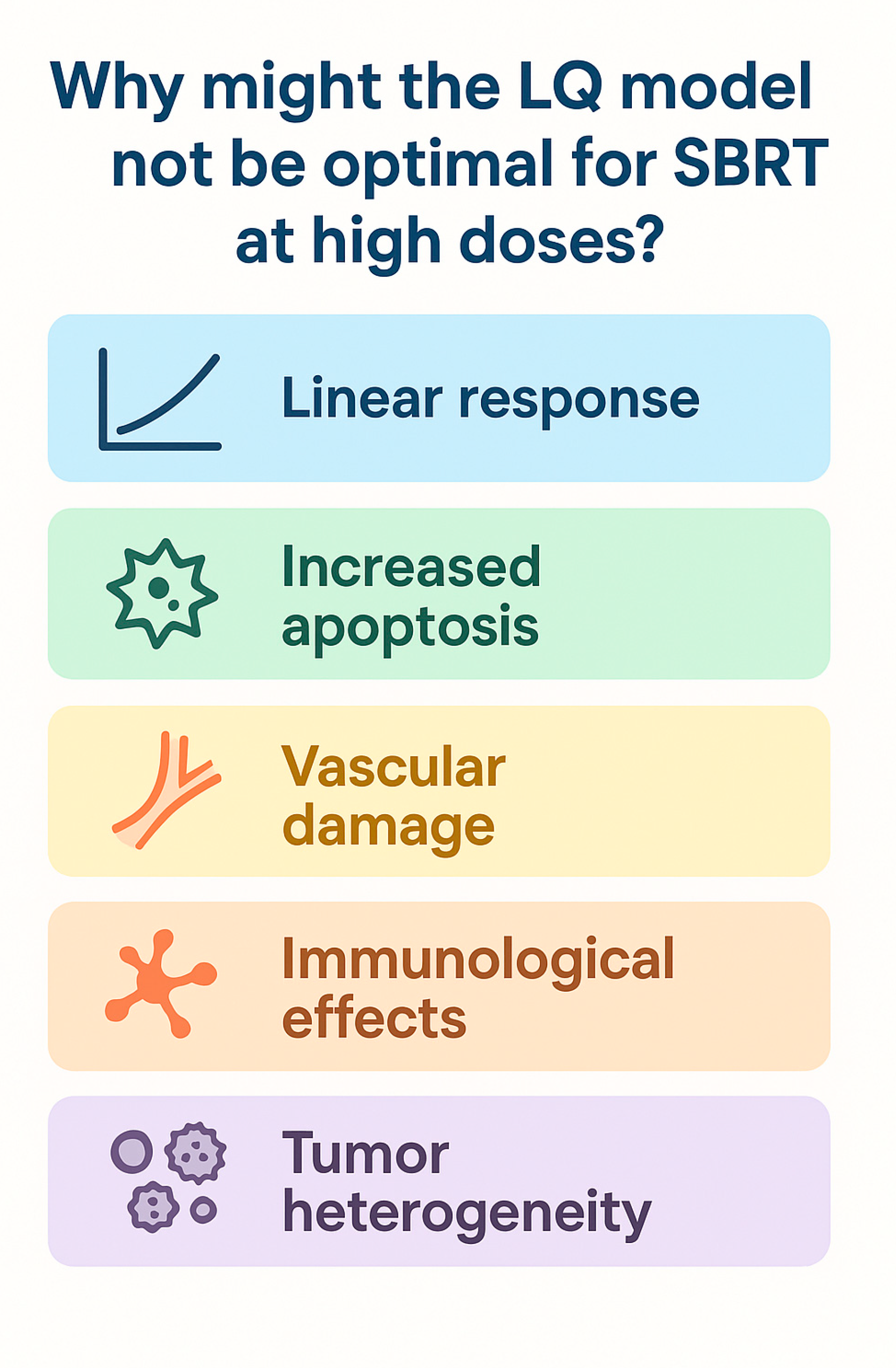
2.2. Limitations of the LQ Model and Treatment Delivery Impact
3. Genetic Factors and Stereotactic Body Radiation Therapy Resistance
3.1. DNA Damage Repair Upregulation
3.2. Tumor Suppressor Mutations and Clonal Selection
3.3. Inflammatory Signaling
4. Genetic Determinants of SBRT Resistance in Solid Tumors
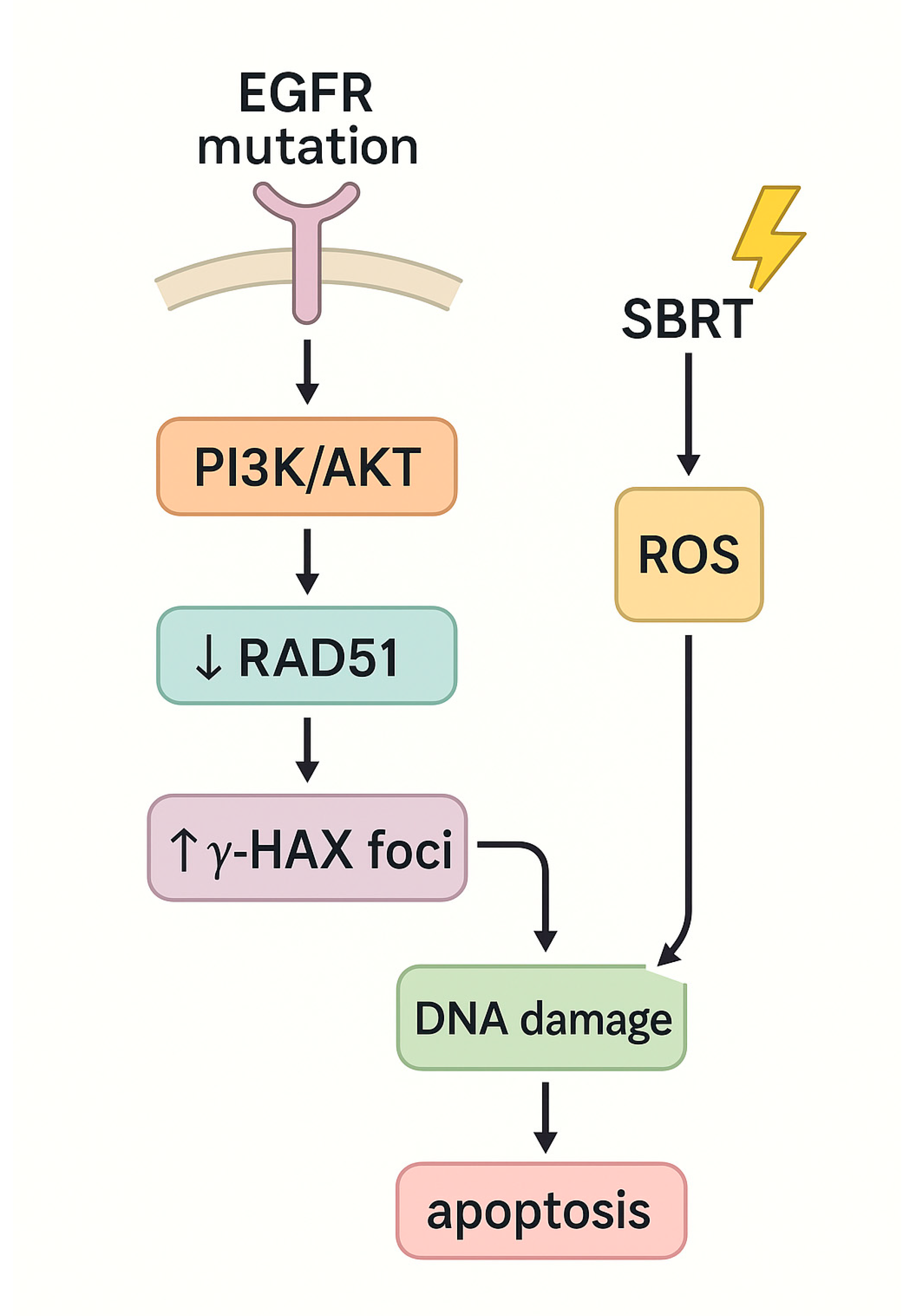
4.1. Non-Small Cell Lung Cancer (NSCLC)
4.2. Pancreatic Ductal Adenocarcinoma (PDAC)
4.3. Renal Cell Carcinoma (RCC)
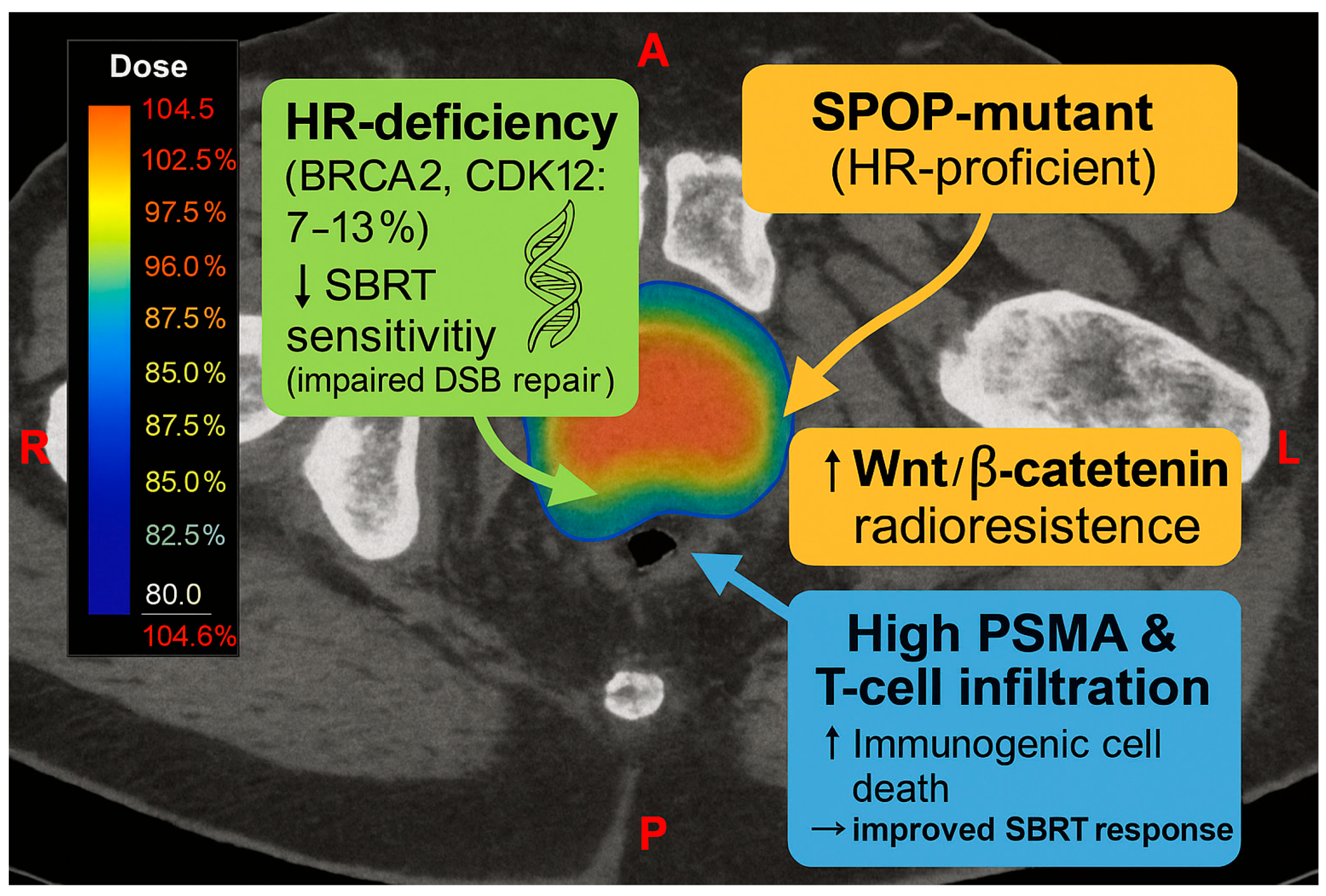
4.4. Prostate Cancer
- •
- Overexpression of DNA-PKcs, which promotes efficient DSB repair via NHEJ.
- •
- Constitutive activation of ATM/ATR signaling, which facilitates prolonged cell cycle arrest and damage repair
- •
4.5. Other Solid Tumors
5. Discussion
- Prediction of SBRT resistance: Radiogenomic profiling can identify tumors harboring mutations (e.g., in TP53, KEAP1) or gene expression patterns (e.g., high PRKDC or RAD51) predictive of poor response. Models such as Genomic-Adjusted Radiation Dose (GARD) use gene expression to estimate the biologic effect of radiation and suggest when dose escalation or combination therapy may be warranted [29].
- Targeted radiosensitization strategies: Tumors with DDR upregulation may benefit from ATM, ATR, or DNA-PK inhibitors. Preclinical successes (e.g., ATR inhibitors in pancreatic cancer, ATM inhibitors in glioma) are now entering clinical trials [24,56]. In tumors with impaired apoptosis (e.g., TP53 or SMAD4 mutants), combining SBRT with BH3 mimetics or PARP inhibitors may induce synthetic lethality [64].
- Integration with immunotherapy: Tumors with high tumor mutation burden or intact STING signaling may benefit from SBRT-induced immune activation. Conversely, tumors with immune-evasive genotypes (e.g., KEAP1, LKB1) may require metabolic or inflammatory modulators (e.g., glutaminase inhibitors) in combination with SBRT and immunotherapy [13,17,33,36].
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
List of Abbreviations
| BED | Biologically Effective Dose | |
| CAF | Cancer-Associated Fibroblast | |
| CRC | Colorectal Cancer | |
| DDR | DNA Damage Response | |
| DNA-PK | DNA-Dependent Protein Kinase | |
| DSB | Double-Strand Break | |
| EGFR-TKI | Epidermal Growth Factor Receptor–Tyrosine Kinase Inhibitor | |
| GARD | Genomic-Adjusted Radiation Dose | |
| GWAS | Genome-Wide Association Studies | |
| HCC | Hepatocellular Carcinoma | |
| HR | Homologous Recombination | |
| IL-6 | Interleukin-6 | |
| LAPC | Locally Advanced Pancreatic Cancer | |
| LQ | Linear-Quadratic model | |
| MMR | Mismatch Repair | |
| NF-κB | Nuclear Factor kappa B | |
| NHEJ | Non-Homologous End-Joining | |
| NSCLC | Non-Small Cell Lung Cancer | |
| PARP | Poly(ADP-ribose) Polymerase | |
| PDAC | Pancreatic Ductal Adenocarcinoma | |
| PSMA | Prostate-Specific Membrane Antigen | |
| RCC | Renal Cell Carcinoma | |
| ROS | Reactive Oxygen Species | |
| SABR | Stereotactic Ablative Radiotherapy | |
| SBRT | Stereotactic Body Radiotherapy | |
| SRS | Stereotactic Radiosurgery | |
| TGF-β | Transforming Growth Factor-beta | |
| TNF-α | Tumor Necrosis Factor-alpha | |
| VEGF | Vascular Endothelial Growth Factor | |
| Genes | ||
| ATM | Ataxia Telangiectasia Mutated | |
| ATR | ATM and Rad3-Related | |
| BRCA1 | Breast Cancer 1 | |
| BRCA2 | Breast Cancer 2 | |
| EGFR | Epidermal Growth Factor Receptor | |
| KEAP1 | Kelch-Like ECH-Associated Protein 1 | |
| KRAS | Kirsten Rat Sarcoma Viral Oncogene Homolog | |
| LKB1 | Liver Kinase B1 | |
| PTEN | Phosphatase and Tensin Homolog | |
| RAD51 | RAD51 Recombinase | |
| SMAD4 | SMAD Family Member 4 | |
| TP53 | Tumor Protein 53 | |
| VHL | Von Hippel–Lindau | |
References
- West, C.M.; Barnett, G.C. Genetics and genomics of radiotherapy toxicity: Towards prediction. Genome Med. 2011, 3, 52. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Shen, L. Radiobiology of stereotactic ablative radiotherapy (SABR): Perspectives of clinical oncologists. J. Cancer 2020, 11, 5056–5068. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.; Borghesi, S.; Aristei, C.; Becherini, C. Radiobiology of stereotactic radiotherapy. Rep. Pract.Oncol. Radiother. 2022, 27, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kim, W.; Park, I.H.; Kim, H.J.; Lee, E.; Jung, J.-H.; Cho, L.C.; Song, C.W. Radiobiological mechanisms of stereotactic body radiation therapy and stereotactic radiation surgery. Radiat. Oncol. J. 2015, 33, 265–275. [Google Scholar] [CrossRef]
- Bernstein, M.B.; Krishnan, S.; Hodge, J.W.; Chang, J.Y. Immunotherapy and stereotactic ablative radiotherapy (ISABR): A curative approach? Nat. Rev. Clin. Oncol. 2016, 13, 516–524. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhuang, H. Effect of stereotactic radiotherapy on immune microenvironment of lung cancer. Front. Immunol. 2022, 13, 1025872. [Google Scholar] [CrossRef]
- Spiotto, M.; Fu, Y.X.; Weichselbaum, R.R. The intersection of radiotherapy and immunotherapy: Mechanisms and clinical implications. Sci. Immunol. 2016, 1, EAAG1266. [Google Scholar] [CrossRef]
- Rubini, D.; Gagliardi, F.; Menditti, V.S.; D’aMbrosio, L.; Gallo, P.; D’oNofrio, I.; Pisani, A.R.; Sardaro, A.; Rubini, G.; Cappabianca, S.; et al. Genetic profiling in radiotherapy: A comprehensive review. Front. Oncol. 2024, 14, 1337815. [Google Scholar] [CrossRef]
- Harary, P.M.; Rajaram, S.; Chen, M.S.; Hori, Y.S.; Park, D.J.; Chang, S.D. Genomic predictors of radiation response: Recent progress towards personalized radiotherapy for brain metastases. Cell Death Discov. 2024, 10, 501. [Google Scholar] [CrossRef] [PubMed]
- i Garau, M.M. Radiobiology of stereotactic body radiation therapy (SBRT). Rep. Pract. Oncol. Radiother. 2017, 22, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Aili, A.; Xue, L.; Jiang, P.; Wang, J. Advances in Radiobiology of Stereotactic Ablative Radiotherapy. Front. Oncol. 2020, 10, 1165. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Preuss, K.; Milano, M.T.; He, X.; Gou, L.; Shi, Y.; Marples, B.; Wan, R.; Yu, H.; Du, H.; et al. Mathematical modeling in radiotherapy for cancer: A comprehensive narrative review. Radiat. Oncol. 2025, 20, 49. [Google Scholar] [CrossRef]
- Beckers, C.; Pruschy, M.; Vetrugno, I. Tumor hypoxia and radiotherapy: A major driver of resistance even for novel radiotherapy modalities. Semin. Cancer Biol. 2024, 98, 19–30. [Google Scholar] [CrossRef]
- Brown, J.M.; Carlson, D.J.; Brenner, D.J. The tumor radiobiology of SRS and SBRT: Are more than the 5 Rs involved? Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 254–262. [Google Scholar] [CrossRef]
- Sheu, T.; Molkentine, J.; Transtrum, M.K.; Buchholz, T.A.; Withers, H.R.; Thames, H.D.; Mason, K.A. Use of the LQ model with large fraction sizes results in underestimation of isoeffect doses. Radiother. Oncol. 2013, 109, 21–25. [Google Scholar] [CrossRef]
- Wang, J.Z.; Huang, Z.; Lo, S.S.; Yuh, W.T.; Mayr, N.A. A generalized linear-quadratic model for radiosurgery, stereotactic body radiation therapy, and high-dose rate brachytherapy. Sci. Transl. Med. 2010, 2, 39–48. [Google Scholar] [CrossRef]
- Shibamoto, Y.; Miyakawa, A.; Otsuka, S.; Iwata, H. Radiobiology of hypofractionated stereotactic radiotherapy: What are the optimal fractionation schedules? J. Radiat. Res. 2016, 57 (Suppl. S1), i76–i82. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wang, Y.; Tang, J.; Cao, M. Radiotherapy induced immunogenic cell death by remodeling tumor immune microenvironment. Front. Immunol. 2022, 13, 1074477. [Google Scholar] [CrossRef]
- Durante, M.; Formenti, S.C. Radiation-Induced Immunogenic Cell Death and Abscopal Effects in Cancer Therapy. Nat. Rev. Cancer 2020, 20, 360–370. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, W.; Li, N.; Neri, S.; Sharma, A.; Jiang, W.; Lin, S.H. Combining Immunotherapy and Radiotherapy for Cancer Treatment: Current Challenges and Future Directions. Front. Pharmacol. 2018, 9, 185. [Google Scholar] [CrossRef]
- Yang, L.; Shen, C.; Estrada-Bernal, A.; Robb, R.; Chatterjee, M.; Sebastian, N.; Webb, A.; Mo, X.; Chen, W.; Krishnan, S.; et al. Oncogenic KRAS Drives Radioresistance Through Upregulation of NRF2-53BP1-Mediated Non-homologous End-Joining Repair. Nucleic Acids Res. 2021, 49, 11067–11082. [Google Scholar] [CrossRef]
- Bian, L.; Meng, Y.; Zhang, M.; Guo, Z.; Liu, F.; Zhang, W.; Ke, X.; Su, Y.; Wang, M.; Yao, Y.; et al. ATM Expression Is Elevated in Established Radiation-Resistant Breast Cancer Cells and Improves DNA Repair Efficiency. Int. J. Biol. Sci. 2020, 16, 1096–1106. [Google Scholar] [CrossRef]
- Skinner, H.D.; Sandulache, V.C.; Ow, T.J.; Meyn, R.E.; Yordy, J.S.; Beadle, B.M.; Fitzgerald, A.L.; Giri, U.; Ang, K.K.; Myers, J.N. TP53 Disruptive Mutations Lead to Head and Neck Cancer Treatment Failure Through Inhibition of Radiation-Induced Senescence. Clin. Cancer Res. 2012, 18, 290–300. [Google Scholar] [CrossRef]
- Wang, F.; Yu, T.; Zhao, Z.; Li, X.; Song, Y.; He, Y.; Zhou, Y.; Li, P.; An, L.; Wang, F. SMAD4 Limits PARP1-Dependent DNA Repair to Render Pancreatic Cancer Cells Sensitive to Radiotherapy. Cell Death Dis. 2024, 15, 818. [Google Scholar] [CrossRef]
- Wu, C.T.; Chen, M.F.; Chen, W.C.; Hsieh, C.C. The Role of IL-6 in the Radiation Response of Prostate Cancer. Radiat. Oncol. 2013, 8, 159. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, L.; Yang, M.; Wu, X.; Wang, X.; Huang, W.; Yuan, L.; Pan, H.; Wang, Y.; Wang, Z.; et al. Cancer-Associated Fibroblasts Promote the Survival of Irradiated Nasopharyngeal Carcinoma Cells via the NF-κB Pathway. J. Exp. Clin. Cancer Res. 2021, 40, 87. [Google Scholar] [CrossRef] [PubMed]
- Sitthideatphaiboon, P.; Galan-Cobo, A.; Negrao, M.V.; Qu, X.; Poteete, A.; Zhang, F.; Liu, D.D.; Lewis, W.E.; Kemp, H.N.; Lewis, J.; et al. STK11/LKB1 Mutations in NSCLC Are Associated with KEAP1/NRF2-Dependent Radiotherapy Resistance and Targetable by Glutaminase Inhibition. Clin. Cancer Res. 2021, 27, 1720–1733. [Google Scholar] [CrossRef] [PubMed]
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.-F.; Cai, L.; Zheng, D.; et al. Androgen Receptor Signaling Regulates DNA Repair in Prostate Cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.G.; Sedor, G.; Ellsworth, P.; A Scarborough, J.; A Ahmed, K.; E Oliver, D.; A Eschrich, S.; Kattan, M.W.; Torres-Roca, J.F. Pan-Cancer Prediction of Radiotherapy Benefit Using Genomic-Adjusted Radiation Dose (GARD): A Cohort-Based Pooled Analysis. Lancet Oncol. 2021, 22, 1221–1229. [Google Scholar] [CrossRef]
- de Mey, S.; Dufait, I.; De Ridder, M. Radioresistance of Human Cancers: Clinical Implications of Genetic Expression Signatures. Front. Oncol. 2021, 11, 761901. [Google Scholar] [CrossRef]
- Ji, Y.; Zhang, Y.; Liu, S.; Li, J.; Jin, Q.; Wu, J.; Duan, H.; Liu, X.; Yang, L.; Huang, Y. The epidemiological landscape of lung cancer: Current status, temporal trend and future projections based on the latest estimates from GLOBOCAN 2022. J. Natl. Cancer Cent. 2025, in press. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Buchberger, D.S.; Videtic, G.M. Stereotactic Body Radiotherapy for the Management of Early-Stage Non–Small-Cell Lung Cancer: A Clinical Overview. JCO Oncol. Pract. 2023, 19, 239–249. [Google Scholar] [CrossRef]
- Videtic, G.M.M. Stereotactic Body Radiation Therapy Lung Cancer; Simon, L., Bin, T., Nina, M., et al., Eds.; Future Medicine Ltd.: London, UK, 2013; pp. 5–19. [Google Scholar]
- Videtic, G.M.M.; Stephans, K.L. The role of stereotactic body radiotherapy in the management of non-small cell lung cancer: An emerging standard for the medically inoperable patient? Curr. Oncol. Rep. 2010, 12, 235–241. [Google Scholar] [CrossRef]
- Ouyang, W.; Yu, J.; Nuerjiang, S.; Li, Z.; Wang, D.; Wang, X.; Zhang, J.; Xie, C. Stereotactic body radiotherapy improves the survival of patients with oligometastatic non-small cell lung cancer. Cancer Med. 2019, 8, 4605–4614. [Google Scholar] [CrossRef]
- Scalera, S.; Mazzotta, M.; Cortile, C.; Krasniqi, E.; De Maria, R.; Cappuzzo, F.; Ciliberto, G.; Maugeri-Saccà, M. KEAP1-mutant NSCLC: The catastrophic failure of a cell-protecting hub. J. Thorac. Oncol. 2022, 17, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Hellyer, J.A.; Padda, S.K.; Diehn, M.; Wakelee, H.A. Clinical Implications of KEAP1-NFE2L2 Mutations in NSCLC. J. Thorac. Oncol. 2021, 16, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Arolt, C.; Dugan, M.; Wild, R.; Richartz, V.; Holz, B.; Scheel, A.H.; Brägelmann, J.; Wagener-Ryczek, S.; Merkelbach-Bruse, S.; Wolf, J.; et al. KEAP1/NFE2L2 Pathway Signature Outperforms KEAP1/NFE2L2 Mutation Status and Reveals Alternative Pathway-Activating Mutations in NSCLC. J. Thorac. Oncol. 2023, 18, 1550–1567. [Google Scholar] [CrossRef]
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.-H.; Boroughs, L.K.; Rodriguez, M.L.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef]
- Nadal, E.; Palmero, R.; Muñoz-Pinedo, C. Mutations in the antioxidant KEAP1/NRF2 pathway define an aggressive subset of NSCLC resistant to conventional treatments. J. Thorac. Oncol. 2019, 14, 1881–1883. [Google Scholar] [CrossRef]
- Binkley, M.S.; Jeon, Y.J.; Nesselbush, M.; Moding, E.J.; Nabet, B.Y.; Almanza, D.; Kunder, C.; Stehr, H.; Yoo, C.H.; Rhee, S.; et al. KEAP1/NFE2L2 Mutations Predict Lung Cancer Radiation Resistance That Can Be Targeted by Glutaminase Inhibition. Cancer Discov. 2020, 10, 1826–1841. [Google Scholar] [CrossRef]
- Skoulidis, F.; Byers, L.A.; Diao, L.; Papadimitrakopoulou, V.A.; Tong, P.; Izzo, J.; Behrens, C.; Kadara, H.; Parra, E.R.; Canales, J.R.; et al. Co-occurring Genomic Alterations Define Major Subsets of KRAS-Mutant Lung Adenocarcinoma with Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities. Cancer Discov. 2015, 5, 860–877. [Google Scholar] [CrossRef]
- Harris, E.; Thawani, R. Current perspectives of KRAS in non-small cell lung cancer. Curr. Probl. Cancer 2024, 51, 0147–0272. [Google Scholar] [CrossRef]
- El Osta, B.; Behera, M.; Kim, S.; Berry, L.D.; Sica, G.; Pillai, R.N.; Owonikoko, T.K.; Kris, M.G.; Johnson, B.E.; Kwiatkowski, D.J.; et al. Characteristics and Outcomes of Patients with Metastatic KRAS-Mutant Lung Adenocarcinomas: The Lung Cancer Mutation Consortium Experience. J. Thorac. Oncol. 2019, 25, 876–889. [Google Scholar] [CrossRef]
- Zhu, D.Q.; Liu, Y.; Yu, Z.J.; Zhang, R.H.; Li, A.W.; Gong, F.Y.; Wang, W.; Xiao, W.; Fan, Q. The Diverse Analysis Identifies Mutated KRAS Associated with Radioresistance in Non-Small Cell Lung Cancer. World J. Oncol. 2022, 13, 84–95. [Google Scholar] [CrossRef]
- Mogi, A.; Kuwano, H. TP53 mutations in nonsmall cell lung cancer. J. Biomed. Biotechnol. 2011, 2011, 583929. [Google Scholar] [CrossRef]
- Ahrendt, S.A.; Hu, Y.; Buta, M.; McDermott, M.P.; Benoit, N.; Yang, S.C.; Wu, L.; Sidransky, D. p53 mutations and survival in stage I non-small-cell lung cancer: Results of a prospective study. J. Natl. Cancer Inst. 2003, 95, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Shor, D.; Zeng, K.; Chen, H.; Louie, A.; Menjak, I.; Atenafu, E.; Tseng, C.; Detsky, J.; Larouche, J.; Zhang, B.; et al. Molecular Status Predicts for Local Control in Patients with Non-Small Cell Lung Cancer Spinal Metastases Following Spine Stereotactic Body Radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2023, 117, e57–e58. [Google Scholar] [CrossRef]
- Kuruvilla, M.S.; Liu, G.; Syed, I.; Gwadry-Sridhar, F.; Sheffield, B.S.; Sachdeva, R.; Pencz, A.; Zhan, L.; Hueniken, K.; Patel, D.; et al. EGFR mutation prevalence, real-world treatment patterns, and outcomes among patients with resected, early-stage, non-small cell lung cancer in Canada. Lung Cancer 2022, 173, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic Cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Neibart, S.S.; Moningi, S.; Jethwa, K.R. Stereotactic Body Radiation Therapy for Locally Advanced Pancreatic Cancer. Clin. Exp. Gastroenterol. 2024, 17, 213–225. [Google Scholar] [CrossRef]
- Hu, B.; Ma, X.; Huang, R.; Wu, Z.; Lu, J.; Guo, Y.; Tang, J.; Ma, C.; Ma, J.; Zhang, L.; et al. Identification of Key Genes Mutations Associated with the Radiosensitivity by Whole Exome Sequencing in Pancreatic Cancer. Front. Oncol. 2021, 11, 697308. [Google Scholar] [CrossRef]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer Statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef]
- Blackford, A.; Serrano, O.K.; Wolfgang, C.; Parmigiani, G.; Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Eshleman, J.R.; et al. SMAD4 Gene Mutations Are Associated with Poor Prognosis in Pancreatic Cancer. Clin. Cancer Res. 2009, 15, 4674. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xia, X.; Yang, C.; Shen, J.; Mai, J.; Kim, H.-C.; Kirui, D.; Kang, Y.; Fleming, J.B.; Koay, E.J.; et al. SMAD4 Gene Mutation Renders Pancreatic Cancer Resistance to Radiotherapy through Promotion of Autophagy. Clin. Cancer Res. 2018, 24, 3176–3185. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.L.; Pitter, K.L.; Wexler, L.H.; Slotkin, E.K.; Gupta, G.P.; Wolden, S.L. TP53 Mutations Increase Radioresistance in Rhabdomyosarcoma and Ewing Sarcoma. Br. J. Cancer 2021, 125, 576–581. [Google Scholar] [CrossRef]
- Maddalena, M.; Mallel, G.; Nataraj, N.B.; Shreberk-Shaked, M.; Hassin, O.; Mukherjee, S.; Arandkar, S.; Rotkopf, R.; Kapsack, A.; Lambiase, G.; et al. TP53 Missense Mutations in PDAC Are Associated with Enhanced Fibrosis and an Immunosuppressive Microenvironment. Proc. Natl. Acad. Sci. USA 2021, 118, e2025631118. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Yang, L.V.; Abrams, S.L.; Steelman, L.S.; Follo, M.Y.; Cocco, L.; Ratti, S.; Martelli, A.M.; Augello, G.; Cervello, M. Effects of TP53 Mutations and miRs on Immune Responses in the Tumor Microenvironment Important in Pancreatic Cancer Progression. Cells 2022, 11, 2155. [Google Scholar] [CrossRef]
- Fan, Z.; Fan, K.; Yang, C.; Huang, Q.; Gong, Y.; Cheng, H.; Jin, K.; Liu, C.; Ni, Q.; Yu, X.; et al. Critical Role of KRAS Mutation in Pancreatic Ductal Adenocarcinoma. Transl. Cancer Res. 2018, 7. [Google Scholar] [CrossRef]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRASG12D Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef]
- Wang, T.; Chen, J.R.; Pagan, V.B.; Wang, L.; Guo, Y.; Xia, L.; Singh, P.K.; Jiang, D.; Koong, A. Abstract B032: Concurrent Mutant KRAS Inhibition and Stereotactic Body Radiation Therapy (SBRT) for Preclinical KRASG12D-Driven Pancreatic Ductal Adenocarcinoma (PDAC) Treatment. Clin. Cancer Res. 2025, 31 (Suppl. S2), B032. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Shultz, C.W.; Azimi-Sadjadi, A.; Brody, J.R.; Pishvaian, M.J. ATM Dysfunction in Pancreatic Adenocarcinoma and Associated Therapeutic Implications. Mol. Cancer Ther. 2019, 18, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.; Ziranu, P.; Spanu, D.; Dubois, M.; Pretta, A.; Tolu, S.; Camera, S.; Liscia, N.; Mariani, S.; Persano, M.; et al. BRCA-Mutant Pancreatic Ductal Adenocarcinoma. Br. J. Cancer 2021, 125, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- Dardare, J.; Witz, A.; Betz, M.; François, A.; Lamy, L.; Husson, M.; Demange, J.; Rouyer, M.; Lambert, A.; Merlin, J.-L.; et al. DDB2 Expression Lights the Way for Precision Radiotherapy Response in PDAC Cells, with or without Olaparib. Cell Death Discov. 2024, 10, 411. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Msaouel, P.; Hara, K.; Choi, H.; Le, V.; Shah, A.Y.; Wang, J.; Jonasch, E.; Choi, S.; Nguyen, Q.; et al. Definitive Radiotherapy in Lieu of Systemic Therapy for Oligometastatic Renal Cell Carcinoma: A Single-Arm, Single-Centre, Feasibility, Phase 2 Trial. Lancet Oncol. 2021, 22, 1732–1739. [Google Scholar] [CrossRef]
- Berger, N.D.; Stanley, F.K.T.; Moore, S.; Goodarzi, A.A. ATM-Dependent Pathways of Chromatin Remodelling and Oxidative DNA Damage Responses. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160283. [Google Scholar] [CrossRef]
- Ren, W.; Xue, B.; Chen, M.; Liu, L.; Zu, X. Low Expression of ATM Indicates a Poor Prognosis in Clear Cell Renal Cell Carcinoma. Clin. Genitourin. Cancer 2019, 17, e433–e439. [Google Scholar] [CrossRef]
- Chan, K.H.; Clavijo, N.D.; Ayala, G.; Hall, R.; Wray, C.; Cen, P. Double Hit in Clear-Cell Renal Cell Carcinoma with Germline Pathogenic ATM Mutation and Somatic VHL Mutation. J. Investig. Med. High. Impact Case Rep. 2024, 12, 23247096241286370. [Google Scholar] [CrossRef]
- Yanagisawa, T.; Urade, M.; Yamamoto, Y.; Furuyama, J. Increased Expression of Human DNA Repair Genes, XRCC1, XRCC3 and RAD51, in Radioresistant Human KB Carcinoma Cell Line N10. Oral. Oncol. 1998, 34, 524–528. [Google Scholar] [CrossRef]
- Liu, Q.-H.; Wang, Y.; Yong, H.-M.; Hou, P.-F.; Pan, J.; Bai, J.; Zheng, J.-N. XRCC1 Serves as a Potential Prognostic Indicator for Clear Cell Renal Cell Carcinoma and Inhibits Its Invasion and Metastasis through Suppressing MMP-2 and MMP-9. Oncotarget 2017, 8, 109382–109392. [Google Scholar] [CrossRef]
- Zhang, W.; Su, Y.; Yue, G.; Zhao, L.; Li, H.; Jia, M.; Wang, Y.; Liu, D.; Wang, H.; Gao, Y. Correlations of SDF-1α and XRCC1 Gene Polymorphisms with the Risk of Renal Cancer Development and Bioinformatics Studies of SDF-1α and XRCC1 and the Prognosis of Renal Cancer. Sci. Rep. 2024, 14, 3367. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Hinoda, Y.; Matsuyama, H.; Tanaka, Y.; Okayama, N.; Suehiro, Y.; Zhao, H.; Urakami, S.; Kawamoto, K.; Kawakami, T.; et al. Polymorphisms of DNA Repair Genes Are Associated with Renal Cell Carcinoma. Biochem. Biophys. Res. Commun. 2006, 342, 1058–1062. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, H.S.; Rübben, H.; Schmid, K.W.; Siffert, W.; Riemann, K. DNA Repair Gene XRCC1 Polymorphisms and Outcome of Renal Cell Carcinoma in Caucasian Patients. Anticancer Res. 2009, 29, 5131–5135. [Google Scholar]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting Renal Cell Carcinoma with a HIF-2 Antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef]
- Meléndez-Rodríguez, F.; Roche, O.; Sanchez-Prieto, R.; Aragones, J. Hypoxia-Inducible Factor 2-Dependent Pathways Driving Von Hippel–Lindau-Deficient Renal Cancer. Front. Oncol. 2018, 8, 214. [Google Scholar] [CrossRef]
- Shirole, N.H.; Kaelin, W.G. VHL and HIF at the Center of RCC Biology. Hematol. Oncol. Clin. N. Am. 2023, 37, 809–825. [Google Scholar] [CrossRef]
- Blanco, A.I.; Teh, B.S.; Amato, R.J. Role of Radiation Therapy in the Management of Renal Cell Cancer. Cancers 2011, 3, 4010–4023. [Google Scholar] [CrossRef]
- Suárez, C.; Vieito, M.; Valdivia, A.; González, M.; Carles, J. Selective HIF2A Inhibitors in the Management of Clear Cell Renal Cancer and Von Hippel–Lindau-Disease-Associated Tumors. Med. Sci. 2023, 11, 46. [Google Scholar] [CrossRef]
- Karanika, S.; Karantanos, T.; Li, L.; Corn, P.G.; Thompson, T.C. DNA Damage Response and Prostate Cancer: Defects, Regulation and Therapeutic Implications. Oncogene 2015, 34, 2815–2822. [Google Scholar] [CrossRef]
- Oshima, M.; Takayama, K.; Yamada, Y.; Kimura, N.; Kume, H.; Fujimura, T.; Inoue, S. Identification of DNA Damage Response-Related Genes as Biomarkers for Castration-Resistant Prostate Cancer. Sci. Rep. 2023, 13, 19602. [Google Scholar] [CrossRef]
- Maekawa, S.; Takata, R.; Obara, W. Molecular Mechanisms of Prostate Cancer Development in the Precision Medicine Era: A Comprehensive Review. Cancers 2024, 16, 523. [Google Scholar] [CrossRef] [PubMed]
- Lukashchuk, N.; Barnicle, A.; Adelman, C.A.; Armenia, J.; Kang, J.; Barrett, J.C.; Harrington, E.A. Impact of DNA Damage Repair Alterations on Prostate Cancer Progression and Metastasis. Front. Oncol. 2023, 13, 1162644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; van Gent, D.C.; Incrocci, L.; van Weerden, W.M.; Nonnekens, J. Role of the DNA Damage Response in Prostate Cancer Formation, Progression and Treatment. Prostate Cancer Prostatic Dis. 2020, 23, 24–37. [Google Scholar] [CrossRef]
- Sutera, P.; Phillips, R.M.; Deek, M.; Ozyigit, G.; Onal, C.; Tran, P.T. The Promise of Metastasis-Directed Therapy for Oligometastatic Prostate Cancer: Going Beneath the Surface with Molecular Imaging. J. Nucl. Med. 2022, 63, 339–341. [Google Scholar] [CrossRef]
- Ahmed, K.A.; Scott, J.G.; Arrington, J.A.; Naghavi, A.O.; Grass, G.D.; Perez, B.A.; Caudell, J.J.; Berglund, A.E.; Welsh, E.A.; Eschrich, S.A.; et al. Radiosensitivity of Lung Metastases by Primary Histology and Implications for Stereotactic Body Radiation Therapy Using the Genomically Adjusted Radiation Dose. J. Thorac. Oncol. 2018, 13, 1121–1127. [Google Scholar] [CrossRef]
- Lazar, A.J.; McLellan, M.D.; Bailey, M.H.; Miller, C.A.; Appelbaum, E.L.; Cordes, M.G.; Fronick, C.C.; Fulton, L.A.; Fulton, R.S.; Mardis, E.R.; et al. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965.e28. [Google Scholar] [CrossRef]
- Kelleher, F.C.; McArthur, G.A. Targeting NRAS in Melanoma. Cancer J. 2012, 18, 132–136. [Google Scholar] [CrossRef]
- Kudchadkar, R.; Paraiso, K.H.T.; Smalley, K.S.M. Targeting Mutant BRAF in Melanoma: Current Status and Future Development of Combination Therapy Strategies. Cancer J. 2012, 18, 124–131. [Google Scholar] [CrossRef]
- Paraiso, K.H.T.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.A.; et al. PTEN Loss Confers BRAF Inhibitor Resistance to Melanoma Cells through the Suppression of BIM Expression. Cancer Res. 2011, 71, 2750–2760. [Google Scholar] [CrossRef]
- Liang, Z.; Xue, C.; Chen, Q.; Li, M.; Li, G.; Feng, H.; Liu, Y.; Liu, X.; Ma, S. Screening of Prognostic Biomarkers for Stereotactic Body Radiation Therapy in Primary Liver Cancer. Dose Response 2022, 20, 15593258221097589. [Google Scholar] [CrossRef]
- Li, J.; Bai, L.; Xin, Z.; Song, J.; Chen, H.; Song, X.; Zhou, J. TERT-TP53 Mutations: A Novel Biomarker Pair for Hepatocellular Carcinoma Recurrence and Prognosis. Sci. Rep. 2025, 15, 3620. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acronym | Full Term | Target Region | Typical Dose/Fraction | Number of Fractions | Common Clinical Indications |
|---|---|---|---|---|---|
| SABR | Stereotactic Ablative Radiotherapy | Extracranial (general term) | >5 Gy | 1–5 | Lung, liver, spine, prostate |
| SBRT | Stereotactic Body Radiotherapy | Extracranial (body) | >5 Gy | 1–5 | Lung, liver, adrenal, spine |
| SRS | Stereotactic Radiosurgery | Intracranial (brain/spine) | 12–24 Gy (single fraction) | 1 (occasionally 2–5) | Brain metastases, AVMs, vestibular schwannomas |
| Gene | Frequency in PDAC | Mechanism of Radioresistance | Potential Inhibitor/Treatment |
|---|---|---|---|
| SMAD4 | ~55% | ↑ ROS, ↑ autophagy, ↓ PARP1-mediated DNA repair | PARP inhibitors (e.g., olaparib) + RT |
| TP53 | ~75% | ↓ apoptosis, ↑ inflammation, and immune suppression | MDM2 inhibitors (e.g., Nutlin-3a) + RT |
| KRAS | >90% | ↑ NRF2/53BP1-mediated NHEJ, prevention of mitotic catastrophe | KRASG12C inhibitors (e.g., MRTX1133) + SBRT |
| ATM | 2–18% (somatic) | Switch to alternative DNA repair pathways | ATR/PARP inhibitors |
| BRCA1/2 | 3–10% | ↑ TMB, homologous recombination deficiency | Platinum-based chemotherapy, PARP inhibitors |
| Tumor Histology | Key Genomic Characteristics | Potential Biomarkers |
|---|---|---|
| Soft -tissue sarcomas/melanoma metastases | Sarcomas: complex karyotype, increased expression of DNA repair and cell-cycle genes; Melanoma: BRAF/NRAS mutations, PTEN loss | Historically “radioresistant”—often require higher SBRT doses for equivalent control |
| Breast cancer metastases | Radiosensitive primary disease | Example of high efficacy with standard SBRT doses |
| Colorectal adenocarcinoma metastases | Intact MMR system, EGFR/AKT pathway activation | Tend toward chemoresistance and radioresistance |
| Hepatocellular carcinoma (HCC) | Common drivers: TP53, CTNNB1, TERT mutations; heterogeneous tumor genomics | ADIPOR1: ↑ post-SBRT in responders (100% sensitivity, 83% specificity) EPB42: expression changes correlate with outcome |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vučinić, D.; Bukovica Petrc, A.-M.; Antončić, I.; Kolak Radojčić, M.; Lekić, M.; Couñago, F. Radiogenomics of Stereotactic Radiotherapy: Genetic Mechanisms Underlying Radiosensitivity, Resistance, and Immune Response. Genes 2025, 16, 732. https://doi.org/10.3390/genes16070732
Vučinić D, Bukovica Petrc A-M, Antončić I, Kolak Radojčić M, Lekić M, Couñago F. Radiogenomics of Stereotactic Radiotherapy: Genetic Mechanisms Underlying Radiosensitivity, Resistance, and Immune Response. Genes. 2025; 16(7):732. https://doi.org/10.3390/genes16070732
Chicago/Turabian StyleVučinić, Damir, Ana-Marija Bukovica Petrc, Ivona Antončić, Maja Kolak Radojčić, Matea Lekić, and Felipe Couñago. 2025. "Radiogenomics of Stereotactic Radiotherapy: Genetic Mechanisms Underlying Radiosensitivity, Resistance, and Immune Response" Genes 16, no. 7: 732. https://doi.org/10.3390/genes16070732
APA StyleVučinić, D., Bukovica Petrc, A.-M., Antončić, I., Kolak Radojčić, M., Lekić, M., & Couñago, F. (2025). Radiogenomics of Stereotactic Radiotherapy: Genetic Mechanisms Underlying Radiosensitivity, Resistance, and Immune Response. Genes, 16(7), 732. https://doi.org/10.3390/genes16070732