1. Introduction
The evolution of eukaryotic genomes is characterized by a dynamic balance between conservation and innovation. At the heart of this process lies the continuous origin of new genes, which provide the raw material for functional diversification, environmental adaptation, and the increase in biological complexity. Traditionally, genes have been considered stable and well-defined entities; however, modern genomic and transcriptomic analyses have revealed a much more fluid and modular landscape, in which gene birth and loss are frequent events, even over relatively short evolutionary timescales.
Throughout evolution, the emergence of new genes has played a fundamental role in shaping the molecular architecture of organisms, contributing to the appearance of novel phenotypic traits and metabolic pathways. These events are particularly relevant in eukaryotes, whose genomes exhibit exon–intron gene structures and a large proportion of non-coding DNA, both of which facilitate molecular innovation. In addition, the high degree of regulatory plasticity, the presence of transposable elements, and the propensity for genomic and sub-genomic duplications make eukaryotic genomes particularly fertile grounds for the emergence of new genes. Recent studies also suggest that nuclear genome organization and chromatin topology contribute to this dynamic, influencing the likelihood that new genetic elements are transcribed, regulated, and functionally retained.
This review aims to systematically explore the main molecular mechanisms involved in the formation of new genes in eukaryotes, with a particular focus on gene duplication, protein domain reshuffling, de novo birth from non-coding regions, and the fixation of these novelties within populations. Emblematic examples of expanding gene families will also be discussed, along with the selective role of functional innovations in mammals and the human species.
2. Mechanisms of Gene Emergence and Divergence
The origin of new genes represents one of the main sources of biological and evolutionary innovation. In eukaryotes, various molecular mechanisms contribute to the formation of novel genes or to the structural rearrangement of existing ones, promoting functional diversification and species adaptation.
2.1. Gene Duplication and Functional Divergence
One of the main mechanisms for the emergence of new genes is the duplication of a pre-existing gene. This process generates two identical, non-allelic copies that can be located either adjacently or distantly within the genome [
1]. While one copy retains the original function, the other is free to accumulate mutations that may alter its function without compromising organismal viability. Evolutionary outcomes of the duplicated gene include neofunctionalization (acquisition of a novel function), subfunctionalization (partitioning of the ancestral function), or pseudogenization (loss of function) [
2].
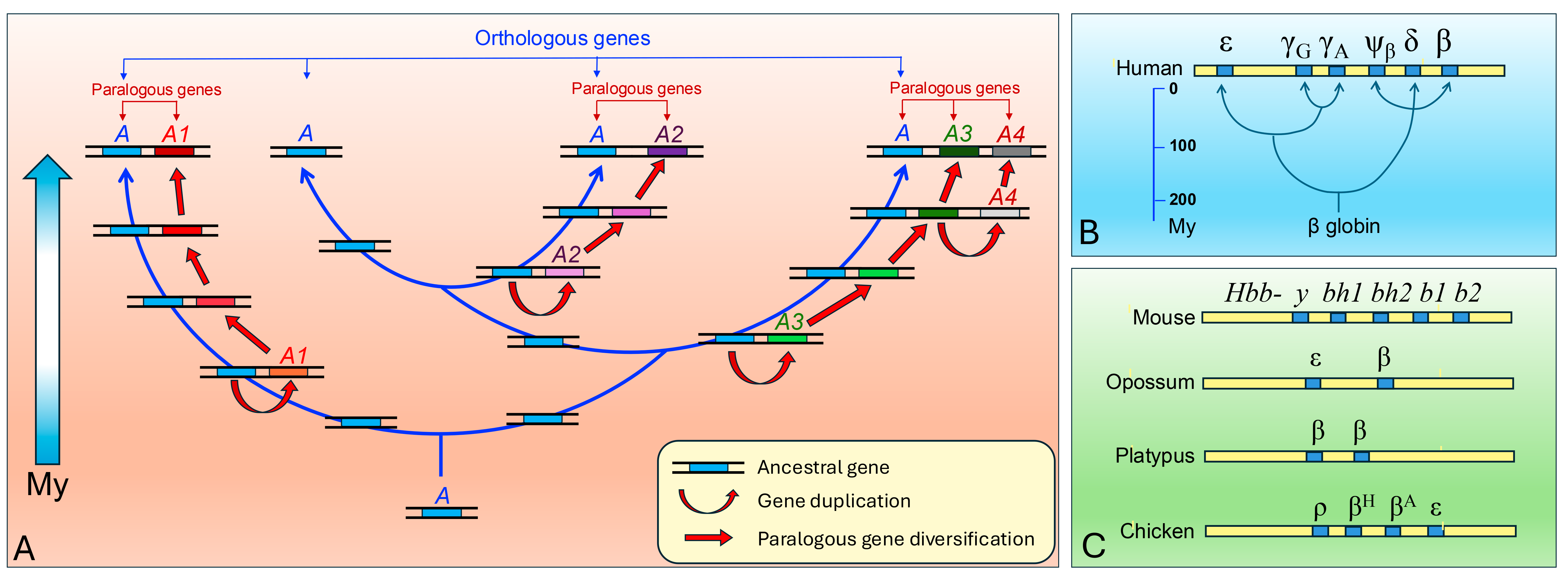
This dynamic has led to the formation of numerous gene families, in which paralogous genes share high sequence homology but perform partially distinct functions (
Figure 1). A typical example is represented by globin genes, where duplication and divergence generated isoforms adapted to different physiological or environmental contexts [
3,
4]. The γ globin subunit of fetal haemoglobin, for instance, exhibits higher oxygen affinity than the adult β globin, optimizing gas exchange across the placenta [
5]. In marine mammals, muscle myoglobin displays unique features that enhance oxygen storage during deep diving [
6]. Similarly, adult hemoglobin’s in high-altitude-adapted species such as llamas exhibit the oxygen affinity properties of fetal forms [
7]. These examples demonstrate how subtle variations in gene sequence can produce substantial biological effects and drive adaptation.
2.2. Exon Duplication
Beyond whole-gene duplication, structural mutations can lead to the formation of new genes through exon duplication or reshuffling, mechanisms facilitated by the modular architecture of eukaryotic genes [
10]. Serial duplication of exons can result in the extension of a gene and the repetition of specific protein domains, potentially conferring novel structural or functional properties.
An emblematic case is the human gene
apo(a), involved in lipid transport, which originated from a duplication of the
plasminogen gene [
11]. During its evolution, the duplicated copy underwent expansion of the Kringle IV domain, encoded by a specific exon, producing variants with up to 50 repeats and marked inter-individual variability. These variants affect cardiovascular risk and illustrate how the expansion of protein domains can significantly influence physiology [
12].
2.3. Retrotransposition
Retrotransposition is a process mediated by mobile elements, in which an mRNA transcript is reverse-transcribed into complementary DNA (cDNA) and reinserted into the genome. Genes arising through this mechanism, known as retrocopies, initially lack introns and native regulatory sequences, but may occasionally acquire functional promoters and become expressed [
13]. Retrotransposition has generated numerous functional genes in mammals, with a higher frequency compared to other vertebrate lineages [
14]. An illustrative example is
PGAM3, a retrocopy of the glycolytic enzyme
PGAM1, which has acquired testis-specific expression and may play a selective role in primate reproduction [
15].
2.4. Gene Fusion and Fission
2.4.1. Gene Fusion
Gene fusion occurs when parts of two or more previously separate genes combine into a single transcriptional unit, leading to the production of a chimeric protein with domains from different origins. This event can be driven by chromosomal rearrangements, such as translocations or deletions, or by transcriptional readthrough followed by selection [
16]. The newly formed gene may acquire novel functions through the juxtaposition of functional domains not previously associated.
In vertebrates, an emblematic example is the
JAZF1-JJAZ1 gene fusion, frequently observed in endometrial stromal tumors, which generates a chimeric transcription factor with altered regulatory properties [
17]. In evolutionary terms, the creation of new protein architectures through fusion events is a key driver of innovation. For instance, the
TRIM5-Cyclophilin A (
TRIMCyp) fusion gene in some New World monkeys confers resistance to HIV-1-like viruses [
18], illustrating how gene fusion can lead to adaptive advantages.
2.4.2. Gene Fission
Conversely, gene fission refers to the splitting of a single ancestral gene into two or more distinct genes. This can occur through deletion or insertion events that interrupt the original gene structure, followed by functional specialization of the derived units [
19]. Although less common than fusion, fission can also contribute to genomic and functional diversification, as observed in some cases of metabolic gene evolution in bacteria and early eukaryotes.
2.5. Exon Shuffling
Exon shuffling is a molecular mechanism through which exons, particularly those encoding protein domains, are recombined between different genes. This process is facilitated by the modular nature of eukaryotic genes and is often mediated by recombination events involving intronic or intergenic regions. The outcome is the generation of new genes with novel combinations of functional domains, which may lead to innovative structural and biochemical properties [
20].
Shuffling events are especially prevalent in metazoans, where complex exon–intron structures and the abundance of recombination-competent elements enhance the plasticity of gene architectures [
21,
22]. An iconic example is the formation of the tissue-type plasminogen activator (
tPA) gene, which combines domains homologous to epidermal growth factor (EGF), fibronectin, and trypsin (
Figure 2). This modular assembly underlies its dual function in fibrinolysis and cell signaling [
23,
24,
25].
The evolutionary potential of exon shuffling lies in its ability to generate proteins with novel domain architectures, often with minimal deleterious impact, since recombination events typically occur within introns, preserving reading frames.
2.6. De Novo Gene Birth
De novo gene birth is the emergence of new protein-coding genes from previously non-coding DNA sequences. Unlike other mechanisms such as duplication or shuffling, which rely on pre-existing genes or gene fragments, de novo gene formation represents a radical innovation, as it involves the recruitment of entirely novel sequences into coding function [
26].
This process typically requires the appearance of an open reading frame (ORF), the acquisition of transcriptional activity, and in some cases, the establishment of a translation initiation context. Although initially controversial, mounting genomic and transcriptomic evidence now supports the frequent and recurrent origin of de novo genes across diverse taxa, including humans [
27,
28].
Many de novo genes show tissue-specific expression and are often enriched in testes, suggesting roles in reproductive biology and potentially in speciation. For example, the human gene
FLJ33706 (now annotated as
C20orf62,
NCBI Ref. Seq. NM_182584.4) is considered a candidate de novo gene, arising from a previously non-coding intergenic region on chromosome 20 and exhibiting primate-specific expression patterns [
29].
Although most de novo genes are short-lived and subject to rapid turnover, a subset may acquire essential functions and be retained by natural selection, highlighting their evolutionary significance.
2.7. Orphan Genes/Taxonomically Restricted Genes
Orphan genes are defined as genes lacking detectable homologues outside their reference taxon. These genes may originate from de novo events or from rapid divergence that obscures homology with other species. Orphan genes are often involved in taxon-specific biological functions, such as development, environmental adaptation, and reproduction [
30].
In
Drosophila, for instance, many orphan genes expressed during neural development or spermatogenesis appear to have evolved rapidly and may play fundamental roles in the evolution of genus-specific traits. These genes, while not conserved across species, contribute to the unique biology of the taxa in which they are found, potentially facilitating adaptations to specific ecological niches or reproductive strategies [
31,
32].
The study of orphan genes is important for understanding the genetic basis of species-specific characteristics and offers insights into how novel genetic innovations can arise and became fixed within populations.
2.8. Horizontal Gene Transfer
Horizontal gene transfer (HGT) refers to the non-vertical transmission of genetic material between organisms, bypassing traditional parent–offspring inheritance. While long recognized as a major evolutionary force in prokaryotes, its occurrence in eukaryotes—especially multicellular ones—has been more controversial and is typically limited to specific contexts. In unicellular eukaryotes, HGT has played a pivotal role in the acquisition of metabolic and stress-response genes, often from bacterial donors. In multicellular lineages, convincing cases of HGT are rarer but include gene acquisitions in bdelloid rotifers, fungi, and even vertebrate genomes [
33,
34].
A notable example in animals is the presence of microbial genes in the genome of the coffee borer beetle
Hypothenemus hampei, which facilitates the digestion of caffeine and reflects a functional adaptation through HGT [
35]. In humans, HGT-derived sequences are more difficult to detect due to strong vertical inheritance and genomic complexity, yet a few candidate events—particularly involving endogenous retroviral sequences—have been proposed to contribute regulatory elements and protein-coding exons [
36,
37,
38]. Although not a widespread mechanism for new gene birth in complex eukaryotes, HGT nonetheless represents a potential source of innovation, particularly in the context of symbiosis interaction or genomic conflict.
2.9. Viral Gene Domestication as a Mechanism of Novel Gene Emergence
In addition to horizontal gene transfer, another important mechanism of gene emergence in mammals is the domestication of viral genes. Unlike canonical HGT events involving genes from bacteria or unicellular eukaryotes, viral gene domestication refers to the stable incorporation and functional co-option of viral sequences—especially those derived from endogenous retroviruses (ERVs)—into the host genome. These events have given rise to novel genes with essential roles in reproduction, development, immunity, and gene regulation [
39].
A well-documented case is the domestication of retroviral envelope genes, known as syncytins, which have acquired key roles in placental development across mammalian lineages [
40]. Other examples include gag-derived genes with regulatory or structural functions and reverse transcriptase domains repurposed in genomic regulation and transposition control. These domesticated elements are now recognized as a source of genetic novelty and evolutionary innovation, often exhibiting tight transcriptional regulation and tissue-specific functions [
41].
This process of viral domestication represents a distinct route of gene emergence, shaped by ancient viral–host interactions and subject to strong selective constraints following functional integration.
2.10. Alternative Splicing
Alternative splicing is a major evolutionary innovation in eukaryotes, allowing a single gene to produce multiple transcripts and proteins. Its progressive diversification has significantly shaped gene function and organismal complexity throughout eukaryotic evolution. Early eukaryotes likely possessed few introns, but intron gain and loss events—particularly in metazoans and plants—have contributed to the structural and functional evolution of genes. The rise in alternative splicing in multicellular organisms enabled the production of multiple isoforms from a single gene, contributing significantly to proteomic complexity without increasing gene number. Notably, vertebrates exhibit extensive alternative splicing, with tissue- and development-specific patterns. This regulatory versatility has been implicated in the emergence of lineage-specific traits and in the evolution of brain complexity in primates [
42].
A paradigmatic example of cell type-specific alternative splicing is provided by the DDX4 (VASA) gene, an evolutionarily conserved RNA helicase expressed in the germline across metazoans. In mammals, DDX4 undergoes alternative splicing generating isoforms with distinct expression patterns in spermatogonia, spermatocytes, and oocytes, modulating RNA metabolism in a stage-specific manner. Comparative analyses in other organisms such as Drosophila, Xenopus, and mammals reveal both conserved and divergent splicing events, suggesting that lineage-specific splicing patterns of germline genes contribute to the evolution of reproductive strategies and fertility mechanisms in eukaryotes [
43,
44].
A variety of molecular mechanisms underlie the birth of new genes in eukaryotes. These processes differ in origin, frequency, and functional consequences, and include gene duplication, de novo gene birth, horizontal gene transfer, and exon shuffling. The following
Table 1 summarizes their main features, representative examples, and evolutionary outcomes.
These mechanisms have operated with varying prevalence across different eukaryotic lineages, contributing to lineage-specific genomic and phenotypic innovations.
Table 2 illustrates how these mechanisms have shaped gene evolution in major eukaryotic groups, providing representative genes and references.
3. Population Fixation
Once originated, a new gene can follow different paths depending on the selective context and the evolutionary forces at play. Most new sequences do not reach fixation and are lost through genetic drift, negative selection, or simple transcriptional inefficiency. However, in some cases, a new gene provides even a minimal selective advantage, sufficient to promote its spread within the population [
58]. The fixation of a new gene—that is, its stable retention and expansion within the species’ genetic pool—depends on several factors.
3.1. Mechanism of Fixation
3.1.1. Positive Selection and Adaptation
Selective pressure is one of the primary drivers of gene fixation. Genes that provide even a minimal adaptive advantage can be rapidly selected, especially in small populations or those exposed to environmental stressors. This has been observed, for example, in genes involved in immune response, adaptation to new diets or environments, or brain development in the human lineage. In some cases, the fixation of a new gene may be accelerated by sexual selection, as observed for many testis-expressed genes that influence fertility or sperm competition. Positive selection promotes the rapid expansion of genes that improve the fitness of the organism [
59,
60].
3.1.2. Contribution of Genetic Drift
In some circumstances, especially in small populations, a neutral or nearly neutral gene can fix even in the absence of selective advantages through genetic drift. This stochastic process can lead to the fixation of “passive” genes, which may later acquire important functions or become subject to selection. Genetic drift can be particularly significant in isolated or small populations, where the effect of random sampling is more pronounced. A gene that initially does not confer a selective advantage may become more common simply by chance, and if it later acquires a useful function, it may become subject to positive selection [
61].
3.2. Biological Processes Facilitating Fixation
3.2.1. Regulatory Context and Transcriptional Compatibility
For a new gene to be functional, it is not enough for it to be transcribed and translated: it is essential that it is expressed in the correct tissues, at the right times, and in a way that is consistent with pre-existing regulatory networks. Many new genes initially emerge as low-expression transcripts, often in the testes or other permissive tissues, where transcriptional activity is less stringent. This favorable environment allows the nascent gene to “test” its functionality with minimal risk to the organism [
3062].
Over time, if the new protein is not deleterious and acquires a beneficial function, regulatory mutations (in enhancers, promoters, or splice sites) may promote more stable or widespread expression. Mutations that enhance the gene’s expression in the correct tissues and at the right times are critical to its evolutionary success.
3.2.2. Interaction with Pre-Existing Gene Networks
A new gene is more likely to fix if it can interact with existing molecular pathways, helping to modulate or enhance them. This functional integration can occur through the recognition of protein partners, incorporation into regulatory complexes, or interaction with RNA. In some cases, new genes act as modulators or regulators of pre-existing processes, even with initially performing redundant or accessory roles. Interaction with existing metabolic pathways or signaling systems increases the likelihood that the gene will be effectively integrated into the biological system, making its long-term retention more probable [
30].
3.2.3. Expansion Through Duplication Events
Even already-fixed genes can undergo further duplication, leading to the amplification or diversification of their function. A gene originally limited to a specific role or tissue expression may give rise to a gene family or paralogous set with diversified regulation and activity. These events contribute to increased functional complexity and adaptive potential. Gene duplication enables the testing of novel functions without compromising the ancestral one [
63]. After duplication, gene copies may follow distinct evolutionary paths: neofunctionalization, with the acquisition of new roles [
1]; subfunctionalization, where the original function is partitioned among duplicates [
64]; or pseudogenization, leading to functional loss. Duplications may occur as tandem, segmental, or even whole-genome duplications, the latter playing a crucial role in early vertebrate evolution [
65]. In plants, whole-genome duplications are particularly common and well tolerated, with both autopolyploidy (duplication within a single species) and allopolyploidy (duplication following hybridization between species) contributing significantly to plant diversification and ecological adaptation [
66,
67]. These mechanisms are evident in gene families such as the globins, which diversified to meet the oxygen transport demands of different developmental stages [
3]. While often beneficial, gene duplication can also cause imbalances in gene dosage, contributing to disorders such as
MECP2 duplication syndrome. Comparative genomics continues to highlight how gene duplication drives evolutionary innovation, shaping both complexity and lineage-specific traits.
3.2.4. Nuclear Architecture and Spatial Genome Organization Context
The spatial organization of the genome within the interphase nucleus has emerged as a key factor influencing not only gene expression patterns but also the evolutionary fate of newly arisen genes. Chromosomal regions located in gene-rich, transcriptionally active bands typically occupy more internal nuclear positions and are associated with euchromatic environments, favoring stable expression and functional integration [
68]. Moreover, evolutionary studies indicate that such structural features—including chromatin topology and band-specific architecture—are conserved across vertebrates [
69,
70].
Recent research suggests that the chromatin and nuclear context in which a new gene emerges can strongly influence its evolutionary trajectory. Genes arising in GC-rich, euchromatic regions are more likely to become functional and fixed, whereas those in AT-rich, heterochromatic regions often remain silenced or are eventually lost [
71,
72,
73]. Lamina-associated domains (LADs), typically located at the nuclear periphery, are enriched in repressive chromatin marks and correlate with low transcriptional activity [
74,
75]. Their association with the nuclear lamina may constrain the activation and retention of nascent gene sequences. During differentiation, however, some genes can reposition from LADs to more central euchromatic regions, acquiring transcriptional competence—a dynamic shift that may affect their long-term integration into functional networks.
This view is reinforced by research showing that gene-dense regions tend to associate with transcriptionally permissive nuclear sub-compartments such as transcription factories and euchromatic neighborhoods [
76,
77], whereas LADs and pericentromeric regions correlate with gene silencing and reduced activity [
78,
79]. These structural constraints may act as selective filters for gene innovation, favoring the emergence and persistence of new genes in spatially accessible and transcriptionally competent domains.
Altogether, these findings underscore the importance of considering nuclear architecture in evolutionary genomics. The interplay among three-dimensional genome organization, chromatin state, and gene emergence highlights a spatial dimension in the dynamics of gene birth and retention—an aspect increasingly relevant in light of single-cell and 3D genomics approaches.
4. Functional Innovation and Evolutionary Advantages
Once fixed in the population, a new gene can undergo various functional destinies depending on its origin, regulatory context, and protein interactions. The main trajectories for the functional evolution of new genes can be summarized as follows.
4.1. Subfunctionalization vs. Neofunctionalization
In the case of gene duplication, the two paralogs can specialize according to two main scenarios:
Subfunctionalization: Each copy maintains part of the original functions, for example, through spatial or temporal diversification of expression. This process is often driven by neutral mutations and may favor the preservation of both duplicated genes [
64]. In this scenario, each copy performs a subset of the ancestral gene’s functions, potentially ensuring that neither copy is lost despite the redundancy in function.
Neofunctionalization: One copy acquires a novel function not present in the ancestral gene, for example, through mutations affecting active sites, functional domains, or regulatory regions [
1,
80]. This process is central to evolutionary innovation, as it enables the emergence of new functions that may confer a selective advantage to the organism.
4.2. Gene Expression and Post-Duplication Regulation
Regulatory divergence represents a primary pathway for innovation: even with identical coding sequences, modifications in promoters, enhancers, or epigenetic modulators can produce different expression patterns, leading to distinct functions in specific tissues or developmental stages [
81,
82]. The plasticity of new genes is particularly evident in rapidly evolving tissues, such as the brain and testes. This regulatory flexibility allows new genes to be adapted to specific biological contexts, enhancing their potential for functional diversification.
4.3. Epigenetic Regulation, Intrinsically Disordered Proteins, and Functional Plasticity
New genes are often subject to dynamic epigenetic regulation that plays a critical role in modulating their expression and evolutionary potential. Following duplication or origination, one gene copy can be temporarily silenced through DNA methylation
or histone modifications, enabling an “incubation period” during which it accumulates beneficial mutations before being reactivated under specific signals or contexts [
30,
83]. This epigenetic flexibility ensures that gene activation remains tightly controlled and context-dependent, preventing deleterious effects while fostering adaptive innovation.
Moreover, many newly originated genes encode intrinsically disordered proteins (IDPs), characterized by the absence of a stable three-dimensional structure. These proteins are highly versatile, capable of interacting with numerous partners and performing moonlighting functions, i.e., multiple unrelated functions within the same cell [
84,
85]. Their structural flexibility complements epigenetic regulation by allowing the encoded proteins to adapt functionally in complex cellular networks, particularly in rapidly evolving tissues such as the brain and immune system.
Together, epigenetic modulation and the intrinsic disorder of many new proteins contribute to a high degree of functional plasticity. This dual mechanism facilitates the fine-tuning of gene expression and broadens the potential biological roles of new genes, promoting their successful integration and retention in the genome under varying environmental or developmental conditions.
5. Emblematic Examples in Vertebrates and Humans
The study of gene origin and diversification has led to the identification of numerous emblematic examples in vertebrates, particularly in humans, where the formation of new genes has played a pivotal role in the evolution of complex biological functions, the emergence of species-specific traits, and susceptibility to diseases. This section aims to highlight some of the most representative cases, with a particular focus on gene origin through duplications, de novo events, regulatory subfunctionalization, and adaptive innovations related to reproduction.
5.1. Expanded Gene Families
One of the clearest pieces of evidence for the origin and diversification of new genes is observed in expanded gene families, typically arising from duplication events followed by subfunctionalization or neofunctionalization. Olfactory receptors (ORs) represent the largest gene family in vertebrates, with humans possessing over 400 functional genes and an equal number of pseudogenes. Their expansion is related to the evolution of sensory strategies across diverse environments, such as olfactory specialization in rodents or the regression of olfactory function in higher primates, which has been offset by the development of trichromatic vision [
86].
The
HOX gene complex, which encodes transcription factors essential for anteroposterior embryonic development, has undergone multiple duplication events throughout vertebrate evolutionary history. The shift from a single cluster in protochordates to four clusters in vertebrates enabled greater modularity and complexity in body development, facilitating the evolution of specialized structures [
87,
88].
Another notable example is the
KRAB-ZNF gene family, which includes over 350 genes in humans. The proteins encoded by these genes contain zinc finger domains coupled with KRAB domains, which mediate a repressive function. This family has expanded particularly in primates, as an evolutionary response to the proliferation of transposable elements, which are repressed through the recruitment of epigenetic cofactors such as TRIM28 [
89].
5.2. Human-Specific Duplicated Genes with Novel Functions
Among the various mechanisms by which new genes originate, one of the most intriguing involves duplicated-derived gene birth, the emergence of protein-coding genes from ancestrally non-coding sequences. Unlike genes that arise through duplication and divergence, de novo genes represent truly novel genetic elements, often deriving from intergenic or intronic regions that gain transcriptional activity and, eventually, open reading frames. Although the functional validation of such genes remains challenging, growing evidence suggests that a subset of de novo genes is expressed in a tissue-specific manner—particularly in the brain—and is implicated in the evolution of the neocortex and advanced cognitive functions.
To illustrate the role of newly emerged genes in shaping species-specific traits, we present a selection of well-characterized, human-specific genes—ARHGAP11B, NOTCH2NL, and SRGAP2C—that exemplify distinct evolutionary origins, including partial and segmental duplications, as well as structural modifications that generate novel functions. These case studies have been chosen for their established contributions to the development and expansion of the human neocortex, highlighting how recent gene duplications and structural innovations can drive neurodevelopmental complexity and cognitive evolution.
ARHGAP11B is a human-specific gene that originated approximately 5 million years ago through a partial duplication of the ancestral gene
ARHGAP11A. A subsequent point mutation introduced a novel splice donor site, resulting in a new coding exon and a truncated protein with distinct functional properties. It is predominantly expressed in basal progenitor cells within the developing human neocortex. Its expression promotes the proliferation of these progenitors, leading to an increased pool of neurons and contributing to the expansion of the neocortical surface area—a hallmark of the human brain. Ectopic expression of
ARHGAP11B in the embryonic mouse neocortex has been shown to increase the number of basal progenitors and induce cortical folding (gyrification), a feature absent in the lissencephalic (smooth) mouse brain. These findings suggest that
ARHGAP11B played a pivotal role in the evolutionary expansion and increased complexity of the human neocortex [
54].
The
NOTCH2NL gene family comprises human-specific paralogs—
NOTCH2NLA,
NOTCH2NLB, and
NOTCH2NLC—derived from recent duplications of the ancestral
NOTCH2 gene. These duplications resulted in genes that retain the first few exons of NOTCH2 and acquire unique C-terminal sequences, leading to novel functional properties. These genes enhance Notch signaling, a pathway crucial for maintaining neural progenitor cells in a proliferative state. By promoting Notch activity,
NOTCH2NL delays the differentiation of neural progenitors into neurons, thereby extending the period of cortical neurogenesis. This prolonged proliferative phase contributes to the increased neuronal output and the expansion of the human neocortex. Variations in the copy number of
NOTCH2NL genes have been associated with neurodevelopmental disorders such as microcephaly and macrocephaly, underscoring their role in brain size regulation. The emergence of
NOTCH2NL is believed to have been a key event in the evolution of the human brain, facilitating the development of its unique structural and functional features [
55].
SRGAP2C is a human-specific paralog of the ancestral
SRGAP2A gene, arising approximately 2.4 million years ago through an incomplete segmental duplication. This duplication resulted in a truncated version of
SRGAP2A that retains the F-BAR domain but lacks the RhoGAP and SH3 domains.
SRGAP2C dimerizes with
SRGAP2A, acting as a dominant-negative inhibitor. This interaction inhibits
SRGAP2A’s role in promoting dendritic spine maturation and limiting spine density. Consequently,
SRGAP2C expression leads to increased spine density and prolonged periods of synaptic development (neoteny), features associated with enhanced synaptic plasticity and cognitive abilities in humans [
90].
These examples illustrate how de novo genes can endow species with unique traits, highlighting their potential to influence complex biological processes such as brain development, neuronal differentiation, and cognitive function. In the human lineage, such genes may have contributed to the emergence of species-specific features, including the expanded neocortex and enhanced synaptic plasticity. Together, these findings underscore the remarkable capacity of new genes to acquire essential roles within a relatively short evolutionary timescale, reshaping our understanding of how genomic novelty drives phenotypic innovation.
5.3. Mammalian-Specific Duplicated Genes: Functional Divergence and Physiological Innovation
5.3.1. Fetal Hemoglobin: Regulatory Subfunctionalization
The
HBG1 and
HBG2 genes, which encode the γ-globin chains of fetal hemoglobin (HbF, α₂γ₂), originated via a duplication event of an ancestral
β-globin gene within the
β-globin gene cluster located on human chromosome 11 (
Figure 1) [
8,
9]. This duplication, which occurred early in the evolution of eutherian mammals, led to the formation of a multi-gene family that includes embryonic, fetal, and adult globin genes arranged sequentially and expressed in a developmentally regulated manner [
91,
92]. During fetal development,
HBG1 and
HBG2 are actively transcribed, producing γ-globin chains that, in combination with α-globin, form fetal hemoglobin. HbF has a higher affinity for oxygen than adult hemoglobin (HbA), allowing efficient oxygen transfer from maternal to fetal circulation—a critical adaptation for intrauterine life [
93]. After birth, a switch in globin gene expression occurs: γ-globin expression is downregulated, and the adult
β-globin gene (
HBB) becomes the predominant transcript, resulting in the formation of HbA (α₂β₂).
This developmental switch is tightly regulated by epigenetic modifications, including DNA methylation and histone modifications, as well as by long-range interactions mediated by the locus control region (LCR) upstream of the
β-globin cluster [
94,
95]. The silencing of
HBG genes postnatally exemplifies a regulatory subfunctionalization process, wherein duplicated genes partition their expression domains—in this case, temporally—enabling fine-tuned physiological adaptation without requiring new coding functions. Moreover, this system represents a striking example of adaptive evolution following gene duplication, where changes in regulatory elements confer selective advantages, such as enhanced oxygen transport in the fetus—an essential trait in viviparous mammals [
96]. The retention of both
HBG genes, each with slightly different promoter sequences, also contributes to nuanced regulatory control and may influence HbF levels in adults, a feature of clinical relevance in disorders like β-thalassemia and sickle cell disease [
97].
5.3.2. Caseins: Reproductive Innovation in Mammals
Caseins (CSN1S1, CSN2, CSN3) are abundant milk proteins that facilitate the efficient transport of calcium, phosphorus, and amino acids to the neonate. Casein genes are specific to mammals and likely originated from duplications of ancestral genes involved in protein secretion.
This gene family has acquired new functions related to neonatal nutrition and is closely linked to the evolution of lactation, a trait exclusive to mammals. Caseins have also developed fine regulation in response to hormonal signals such as prolactin and glucocorticoids, contributing to a highly specialized system for offspring care [
98]. This example illustrates how genetic evolution can contribute to the innovation of entire biological systems, with profound effects on physiology and reproductive behavior.
Caseins belong to the SCPP (secretory calcium-binding phosphoprotein) family, which also includes enamel proteins such as amelogenin (AMEL), ameloblastin (AMBN), and enamelin (ENAM). All of these genes originate from tandem duplications of a common ancestor and are located in clusters on human chromosome 4 [
99].
Specifically, caseins appear to have evolved through two distinct evolutionary paths: the calcium-sensitive caseins (such as αS1-, αS2-, and β-casein) evolved from the
SCPPPQ1 gene, which is expressed in dental tissues and shares exonic structures with caseins, and the calcium-insensitive κ-casein evolved from the
FDCSP gene, also expressed in dental tissues. Both of these genes ultimately derive from the ancestral
ODAM (odontogenic ameloblast-associated) gene, expressed during enamel formation [
100].
This origin suggests that caseins inherited the ability to bind calcium from their dental ancestors. Subsequently, these proteins were co-opted to form micelles in milk, a key innovation for neonatal nutrition in mammals. This represents an emblematic example of evolutionary co-option, where pre-existing genes are adapted to new biological functions.
6. From Origin to Function: Methods for Investigating the Emergence of New Genes
The study of gene origin requires the integration of genomic, transcriptomic, phylogenetic, and functional techniques. Thanks to the rapid development of high-resolution technologies and predictive power, it is now possible to analyze not only the emergence of new genes but also their expression, function, and evolutionary impact.
6.1. Comparative Genomics, Phylogeny, and Synteny
Comparative genomics enables the comparison of genomes across different species to identify orthologous genes, gene duplications, and new acquisitions. Gene phylogeny analysis helps reconstruct the evolutionary tree of genes, while synteny (the conservation of gene order between species) is crucial for recognizing de novo gene origins [
101]. For instance, the human gene
ARHGAP11B, involved in cortical development, is absent in other primates and is located in a syntenic region with the ancestral
ARHGAP11A gene, from which it originated through partial duplication followed by subsequent mutations [
91]. This approach allows for the tracing of gene emergence over evolutionary time and provides insights into the mechanisms that drive innovation.
6.2. Transcriptomics and Ribosome Profiling (Ribo-Seq) for the Identification of New ORFs
RNA sequencing (RNA-seq) enables the identification of unannotated transcripts, potentially coding for new genes. However, mere transcription is not sufficient to establish gene functionality. Ribosome profiling (Ribo-seq) allows for the identification of which transcripts are actually translated, even in the case of small open reading frames (sORFs). Recent studies have shown that many regions previously annotated as non-coding actually produce functional micropeptides involved in cellular regulation [
102]. This approach led to the definition of “proto-genes,” emerging transcripts that may evolve into fully functional genes. Together, these technologies are revolutionizing the study of gene function by allowing us to identify potential novel coding regions that were once considered non-coding.
6.3. Selection Analyses and Statistical Tests
To understand the evolutionary dynamics of genes, tests based on the ratio of non-synonymous to synonymous substitutions (dN/dS) are commonly employed. A dN/dS ratio greater than 1 suggests positive selection, whereas a ratio less than 1 indicates purifying selection. These analyses are typically performed using tools like PAML (Phylogenetic Analysis by Maximum Likelihood) (
http://abacus.gene.ucl.ac.uk/software/paml.html, accessed on 18 May 2025) or HyPhy (Hypothesis testing using phylogenies) (
https://www.hyphy.org/, accessed on 18 May 2025). Moreover, the integration of simulated-based models and Bayesian tests provides a robust estimate of selective pressures. Studies on genes like
SRGAP2C have shown signs of positive selection in the human lineage, consistent with its functional acquisition in the control of neuronal migration [
45]. This highlights how genetic innovations are shaped by natural selection to drive the emergence of new traits.
6.4. Experimental Technologies: CRISPR, In Vitro Models, and Organoids
Experimental approaches are crucial for determining the functional role of new genes. The CRISPR-Cas9 technology enables targeted gene knockout or the introduction of specific mutations to test their effects. In addition to CRISPR, classical gene knockout (KO) methods using embryonic stem (ES) cells have been extensively employed to generate animal models, particularly mice, allowing researchers to study gene function in vivo over developmental stages and adult physiology. These ES cell-based KO models have contributed to a vast accumulation of functional data on gene roles, including many involved in development and disease. In vitro models complement these genetic approaches. In particular, induced pluripotent stem cells (iPSCs) and brain organoids have allowed researchers to simulate human development in the laboratory. For instance, introducing
ARHGAP11B into murine neuronal progenitors using gene editing or overexpression systems induced an expansion similar to that observed in the human cortex, suggesting a causal role in cognitive enhancement [
54]. Brain organoids, derived from iPSCs, enable the exploration of gene functions in three-dimensional neural tissue models, bridging the gap between cell culture and whole-organism studies. This highlights the power of combining gene editing and cellular models to investigate the functional significance of new genes in developmental and evolutionary contexts.
Together, these experimental methods, ranging from traditional KO models using ES cells to cutting-edge CRISPR technology and advanced in vitro systems, enable comprehensive functional analyses of new genes allowing the comprehensive tracking of a new gene’s journey, from its appearance to its fixation in the population and the acquisition of a new biological function.
The following
Table 3 provides an overview of the main technologies currently employed to study novel genes, highlighting their applications and representative examples.
7. Conclusions and Future Perspectives
The study of the origin and evolution of genes is not only a fascinating narrative about the ingenuity of nature but also a powerful lens through which we can understand the foundations of biological complexity and our very identity as a species. Analyzing the mechanisms that generate new genes—from duplication and de novo expression to the reorganization of genomic elements—offers critical insights into how new biological functions and innovative adaptations have developed over the course of evolution [
27,
108].
However, many questions remain unanswered:
- •
What is the actual frequency of de novo gene origin in different taxa, and what is their evolutionary stability?
- •
What traits emerge as a direct consequence of the appearance of new genes, and which ones are indirect effects on pre-existing gene networks?
- •
How can we distinguish between a true new functional gene and a “noisy” transcript lacking biological significance?
In the field of evolutionary biology, these questions touch the core of our understanding of speciation, adaptation, and morpho-functional innovation processes. In the biomedical field, identifying recently originated genes could offer new perspectives on complex diseases, human-specific traits, and even the development of targeted therapies. Some recent human genes, such as
NOTCH2NL or
SRGAP2C, are strongly involved in cortical development, suggesting a direct link between genetic innovation and the evolution of higher cognitive abilities [
45,
109]. Others, like variants of the
apo(a) gene, highlight the intersection between gene duplication and cardiovascular risk, serving as a striking example of the clinical potential impact of evolutionary research [
110].
The integration of comparative genomics, epigenomics, structural biology, and systems biology will be essential for fully exploring the hidden potential in new genes and understanding their actual functional relevance. Next-generation sequencing technologies, genomic editing (such as CRISPR), and analysis in advanced model systems (organoids, humanized animals) are already revolutionizing this field. In particular, organoid-based platforms represent promising tools for dissecting the functional roles of novel genes in a tissue- and cell type-specific context, enabling experimental modeling of human-specific traits and pathologies. These approaches are supported by high-resolution reference datasets of human genetic variation [
111], enabling the testing of functional hypotheses and biological causalities that were previously inaccessible.
An additional promising area for research is the study of nuclear architecture and chromatin spatial organization, which may influence both the expression potential and the evolutionary fate of newly emerged genes. Evidence suggests that genes located in gene-rich chromosomal bands or within chromatin territories localized in the more internal compartment of the cell nucleus are more likely to achieve stable expression and functional integration [
68]. The conserved three-dimensional structure of chromosomal regions involved in evolutionary rearrangements underscores the critical relationship between genome topology and functional innovation, further highlighting how spatial positioning within the nucleus can shape the long-term retention and evolutionary potential of new genes [
70].
A particularly promising direction for future research is the study of the human pangenome, which encompasses the complete genetic diversity within our species, beyond the single reference genome. Traditional genomic approaches have been largely based on a limited number of individuals, often from populations of European ancestry, thus failing to capture the full extent of global human diversity. The pangenome initiative aims to correct this bias by incorporating genomes from multiple individuals representing diverse ancestries, geographic origins, and evolutionary histories. Pangenome analysis is already revealing unexpected levels of structural variation, including insertions, deletions, inversions, and copy number variants, many of which are absent from the current reference genome. Importantly, it is also uncovering previously unannotated genes and novel isoforms, some of which may be population-specific or even individual-specific. To better understand the evolutionary dynamics of these new genetic elements, emerging single-cell multi-omics technologies—combining transcriptomics, epigenomics, and proteomics at cellular resolution—offer the opportunity to reconstruct their lineage-specific expression patterns, regulatory integration, and functional emergence across diverse cell types. These findings have far-reaching implications for both evolutionary biology and medicine. For example, population-specific genes may provide insights into local adaptation, while structural variants may contribute to disease susceptibility or resistance in ways that were previously invisible to reference-based analyses [
112,
113]. This global perspective allows for the rediscovery of new genes, understanding alternative expression patterns, and identifying potentially functional structural variants that were overlooked in classical genomic models.
Future research directions should include the following:
- •
Systematic functional characterization of novel genes in organ-specific or development-specific organoids.
- •
Integration of multi-omics data (transcriptome, epigenome, chromatin topology) at single-cell level to trace gene emergence across cell lineages.
- •
Investigation of how nuclear architecture affects expression potential of lineage-specific genes.
- •
Expansion of functional screens (e.g., CRISPR-based perturbation) targeting unannotated regions discovered through pangenome analysis.
- •
Evaluation of population-specific gene functions in health and disease using comparative organoid or xenograft models.
The pangenome thus represents a key tool for deepening our understanding of the origin and evolution of genes, enhancing the equity of biomedical research by including global human diversity, and improving our ability to interpret the genetic basis of health and disease.
The future of evolutionary genetics will increasingly unfold within an interdisciplinary landscape where genomics, evolutionary biology, computational modeling, developmental biology, and clinical sciences converge. It is precisely at the intersection between evolutionary history, molecular innovation, and clinical application that one of the greatest challenges (and promises) of modern biology lies: understanding not only where we come from but also where we can go.
Author Contributions
Conceptualization, S.S. and C.F.; data curation, S.S., D.B., F.B., E.C., V.S. and C.F.; writing—original draft preparation, S.S., D.B. and C.F.; writing—review and editing, S.S., D.B. and C.F.; supervision, S.S. and C.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
E.C. was supported by a fellowship of the PhD program (University of Catania, Catania, Italy).
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AMBN | Ameloblastin |
| AMEL | Amelogenin gene |
| Apo(a) | Apolipoprotein A |
| ARHGAP11A | Rho GTPase Activating Protein 11A |
| ARHGAP11B | Rho GTPase Activating Protein 11B |
| CSN1S1 | Casein α S1 |
| CSN2 | Casein β |
| CSN3 | Casein Kappa |
| ENAM | Enamelin |
| F-BAR | Fes/CIP4 homology-Bin/Amphiphysin/Rvs (F-BAR) domain proteins |
| FDCSP | Follicular Dendritic Cell Secreted Protein |
| FLJ33706 | Human-specific de novo protein-coding gene (alternative gene symbol C20orf203) |
| HbA | Adult hemoglobin |
| HBB | Hemoglobin Subunit β |
| HbF | Fetal hemoglobin |
| HBG1 | Hemoglobin Subunit γ-1 |
| HBG2 | Hemoglobin Subunit γ 2 |
| HGT | Horizontal gene transfer |
| HOX | homeobox genes |
| IDPs | Intrinsically disordered proteins |
| iPSCs | Induced pluripotent stem cells |
| JAZF- JJAZ1 | Zinc-Finger genes |
| KRAB-ZNF | Krüppel-associated box zinc finger |
| LADs | Lamina-associated domains |
| LCR | Locus Control Region |
| NOTCH2NL | Human-specific gene related to the Notch signaling pathway |
| NOTCH2NLA | Notch 2 N-terminal Like Protein A |
| NOTCH2NLB | Notch 2 N-terminal Like Protein B |
| NOTCH2NLC | Notch 2 N-terminal-Like Protein C |
| ODAM | Odontogenic ameloblast-associated gene |
| ORF | Open Reading Frame |
| Ors | Olfactory receptors |
| PAML | Phylogenetic Analysis by Maximum Likelihood |
| PGAM3 | Phosphoglycerate Mutase Family 3 |
| Ribo-seq | Ribosome profiling |
| RNA-seq | RNA sequencing |
| SCPP | Secretory calcium-binding phosphoproteins |
| SCPPPQ1 | Secretory Calcium-Binding Phosphoprotein Proline-Glutamine Rich 1 |
| SH3 | Src Homology 3 domains |
| sORFs small | Open Reading Frames |
| SRGAP2A | SLIT-ROBO Rho GTPase Activating Protein 2 |
| SRGAP2C | SLIT-ROBO Rho GTPase Activating Protein 2C |
| TRIMCyp | TRIM5-Cyclophilin A |
References
- Ohno, S. Evolution by Gene Duplication; Springer: Berlin/Heidelberg, Germany, 1970; ISBN 978-3-642-86661-6. [Google Scholar]
- Zhang, J. Evolution by gene duplication: An update. Trends Ecol. Evol. 2003, 18, 292–298. [Google Scholar] [CrossRef]
- Hardison, R.C. Evolution of hemoglobin and its genes. Cold Spring Harb. Perspect. Med. 2012, 2, a011627. [Google Scholar] [CrossRef]
- Hoffmann, F.G.; Vandewege, M.W.; Storz, J.F.; Opazo, J.C. Gene Turnover and Diversification of the α- and β-Globin Gene Families in Sauropsid Vertebrates. Genome Biol. Evol. 2018, 10, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Storz, J.F. Hemoglobin-oxygen affinity in high-altitude vertebrates: Is there evidence for an adaptive trend? J. Exp. Biol. 2016, 219, 3190–3203. [Google Scholar] [CrossRef] [PubMed]
- Mirceta, S.; Signore, A.V.; Burns, J.M.; Cossins, A.R.; Campbell, K.L.; Berenbrink, M. Evolution of mammalian diving capacity traced by myoglobin net surface charge. Science 2013, 340, 1234192. [Google Scholar] [CrossRef] [PubMed]
- Hebbel, R.P.; Eaton, J.W.; Kronenberg, R.S.; Zanjani, E.D.; Moore, L.G.; Berger, E.M. Human llamas: Adaptation to altitude in subjects with high hemoglobin oxygen affinity. J. Clin. Investig. 1978, 62, 593–600. [Google Scholar] [CrossRef]
- Opazo, J.C.; Hoffmann, F.G.; Storz, J.F. Genomic evidence for independent origins of β-like globin genes in monotremes and therian mammals. Proc. Natl. Acad. Sci. USA 2008, 105, 1590–1595. [Google Scholar] [CrossRef]
- Philipsen, S.; Hardison, R.C. Evolution of hemoglobin loci and their regulatory elements. Blood Cells Mol. Dis. 2018, 70, 2–12. [Google Scholar] [CrossRef]
- Long, M.; Betrán, E.; Thornton, K.; Wang, W. The origin of new genes: Glimpses from the young and old. Nat. Rev. Genet. 2003, 4, 865–875. [Google Scholar] [CrossRef]
- McLean, J.W.; Tomlinson, J.E.; Kuang, W.J.; Eaton, D.L.; Chen, E.Y.; Fless, G.M.; Scanu, A.M.; Lawn, R.M. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature 1987, 330, 132–137. [Google Scholar] [CrossRef]
- Utermann, G. Genetic architecture and evolution of the lipoprotein(a) trait. Curr. Opin. Lipidol. 1999, 10, 133–141. [Google Scholar] [CrossRef]
- Kaessmann, H.; Vinckenbosch, N.; Long, M. RNA-based gene duplication: Mechanistic and evolutionary insights. Nat. Rev. Genet. 2009, 10, 19–31. [Google Scholar] [CrossRef]
- Marques, A.C.; Dupanloup, I.; Vinckenbosch, N.; Reymond, A.; Kaessmann, H. Emergence of young human genes after a burst of retroposition in primates. PLoS Biol. 2005, 3, e357. [Google Scholar] [CrossRef] [PubMed]
- Féral, C.; Guellaën, G.; Pawlak, A. Human testis expresses a specific poly(A)-binding protein. Nucleic Acids Res. 2001, 29, 1872–1883. [Google Scholar] [CrossRef]
- Rogers, R.L.; Bedford, T.; Hartl, D.L. Formation and longevity of chimeric and duplicate genes in Drosophila melanogaster. Genetics 2009, 181, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Hrzenjak, A.; Moinfar, F.; Tavassoli, F.A.; Strohmeier, B.; Kremser, M.L.; Zatloukal, K.; Denk, H. JAZF1/JJAZ1 gene fusion in endometrial stromal sarcomas: Molecular analysis by reverse transcriptase-polymerase chain reaction optimized for paraffin-embedded tissue. J. Mol. Diagn. 2005, 7, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Sayah, D.M.; Sokolskaja, E.; Berthoux, L.; Luban, J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 2004, 430, 569–573. [Google Scholar] [CrossRef]
- Wang, W.; Yu, H.; Long, M. Duplication-degeneration as a mechanism of gene fission and the origin of new genes in Drosophila species. Nat. Genet. 2004, 36, 523–527. [Google Scholar] [CrossRef]
- Gilbert, W. Why genes in pieces? Nature 1978, 271, 501. [Google Scholar] [CrossRef]
- Patthy, L. Exon shuffling and other ways of module exchange. Matrix Biol. 1996, 15, 301–312. [Google Scholar] [CrossRef]
- Patthy, L. Modular assembly of genes and the evolution of new functions. Genetica 2003, 118, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Ny, T.; Elgh, F.; Lund, B. The structure of the human tissue-type plasminogen activator gene: Correlation of intron and exon structures to functional and structural domains. Proc. Natl. Acad. Sci. USA 1984, 81, 5355–5359. [Google Scholar] [CrossRef]
- Collen, D.; Lijnen, H.R. The tissue-type plasminogen activator story. Arter. Thromb. Vasc. Biol. 2009, 29, 1151–1155. [Google Scholar] [CrossRef]
- Chana-Muñoz, A.; Jendroszek, A.; Sønnichsen, M.; Wang, T.; Ploug, M.; Jensen, J.K.; Andreasen, P.A.; Bendixen, C.; Panitz, F. Origin and diversification of the plasminogen activation system among chordates. BMC Evol. Biol. 2019, 19, 27. [Google Scholar] [CrossRef]
- Carvunis, A.R.; Rolland, T.; Wapinski, I.; Calderwood, M.A.; Yildirim, M.A.; Simonis, N.; Charloteaux, B.; Hidalgo, C.A.; Barbette, J.; Santhanam, B.; et al. Proto-genes and de novo gene birth. Nature 2012, 487, 370–374. [Google Scholar] [CrossRef]
- Knowles, D.G.; McLysaght, A. Recent de novo origin of human protein-coding genes. Genome Res. 2009, 19, 1752–1759. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.D.; Irwin, D.M.; Zhang, Y.P. De novo origin of human protein-coding genes. PLoS Genet. 2011, 7, e1002379. [Google Scholar] [CrossRef]
- Li, C.Y.; Zhang, Y.; Wang, Z.; Zhang, Y.; Cao, C.; Zhang, P.-W.; Lu, S.-J.; Li, X.-M.; Yu, Q.; Zheng, X.; et al. A human-specific de novo protein-coding gene associated with human brain functions. PLoS Comput. Biol. 2010, 6, e1000734. [Google Scholar] [CrossRef] [PubMed]
- Tautz, D.; Domazet-Lošo, T. The evolutionary origin of orphan genes. Nat. Rev. Genet. 2011, 12, 692–702. [Google Scholar] [CrossRef]
- Domazet-Loso, T.; Tautz, D. An evolutionary analysis of orphan genes in Drosophila. Genome Res. 2003, 13, 2213–2219. [Google Scholar] [CrossRef]
- Palmieri, N.; Kosiol, C.; Schlötterer, C. The life cycle of Drosophila orphan genes. eLife 2014, 3, e01311. [Google Scholar] [CrossRef] [PubMed]
- Keeling, P.J.; Palmer, J.D. Horizontal gene transfer in eukaryotic evolution. Nat. Rev. Genet. 2008, 9, 605–618. [Google Scholar] [CrossRef]
- Crisp, A.; Boschetti, C.; Perry, M.; Tunnacliffe, A.; Micklem, G. Expression of multiple horizontally acquired genes is a hallmark of both vertebrate and invertebrate genomes. Genome Biol. 2015, 16, 50. [Google Scholar] [CrossRef]
- Wybouw, N.; Pauchet, Y.; Heckel, D.G.; Van Leeuwen, T. Horizontal Gene Transfer Contributes to the Evolution of Arthropod Herbivory. Genome Biol. Evol. 2016, 8, 1785–1801. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, D.J. Endogenous retroviruses in the human genome sequence. Genome Biol. 2001, 2, reviews1017.1. [Google Scholar] [CrossRef] [PubMed]
- Kaneko-Ishino, T.; Ishino, F. The role of genes domesticated from LTR retrotransposons and retroviruses in mammals. Front. Microbiol. 2012, 3, 262. [Google Scholar] [CrossRef]
- Chuong, E.B.; Rumi, M.A.; Soares, M.J.; Baker, J.C. Endogenous retroviruses function as species-specific enhancer elements in the placenta. Nat. Genet. 2013, 45, 325–329. [Google Scholar] [CrossRef]
- Henriques, T.; Mager, D.L.; Makalowski, W. Evolutionary Dynamics of Endogenous Retroviruses and Their Impact on Mammalian Genomes. Mol. Biol. Evol. 2024, 41, msad278. [Google Scholar] [CrossRef]
- Lavialle, C.; Cornelis, G.; Dupressoir, A.; Esnault, C.; Heidmann, O.; Vernochet, C.; Heidmann, T. Paleovirology of ‘syncytins’, retroviral env genes exapted for a role in placentation. Philos. Trans. R. Soc. B 2013, 368, 20120507. [Google Scholar] [CrossRef]
- Kaneko-Ishino, T.; Ishino, F. Evolution of Mammalian Genomic Imprinting by Retrotransposon Insertions and Domestication. Biomolecules 2023, 13, 150. [Google Scholar] [CrossRef]
- Barbosa-Morais, N.L.; Irimia, M.; Pan, Q.; Xiong, H.Y.; Gueroussov, S.; Lee, L.J.; Slobodeniuc, V.; Kutter, C.; Watt, S.; Colak, R.; et al. The Evolutionary Landscape of Alternative Splicing in Vertebrate Species. Science 2012, 338, 1587–1593. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, E.A.; Wessel, G.M. Vasa genes: Emerging roles in the germ line and in multipotent cells. BioEssays 2010, 32, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Lasko, P. The DEAD-box helicase Vasa: Evidence for a multiplicity of functions in RNA processes and developmental biology. Biochim. Biophys. Acta 2013, 1829, 810–816. [Google Scholar] [CrossRef]
- Charrier, C.; Joshi, K.; Coutinho-Budd, J.; Kim, J.E.; Lambert, N.; de Marchena, J.; Jin, W.; Vanderhaeghen, P.; Ghosh, A.; Sestan, N. Inhibition of SRGAP2 function by its human-specific paralogs induces neoteny during spine maturation. Cell 2012, 149, 923–935. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, E.A.; Meselson, M.; Arkhipova, I.R. Massive horizontal gene transfer in bdelloid rotifers. Science 2008, 320, 1210–1213. [Google Scholar] [CrossRef]
- Patthy, L. Evolution of the proteases of blood coagulation and fibrinolysis by assembly from modules. Cell 1985, 41, 657–663. [Google Scholar] [CrossRef]
- El-Sayed, N.M.A.; Ghedin, E.; Hertz-Fowler, C.; Blandin, G.; Renauld, H.; Bartholomeu, D.C.; Lennard, N.J.; Caler, E.; Hamlin, N.E.; Haas, B.; et al. The genome sequence of Trypanosoma brucei. Science 2005, 309, 416–422. [Google Scholar] [CrossRef]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Maréchal-Drouard, L.; et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 2007, 318, 245–250. [Google Scholar] [CrossRef]
- Srivastava, M.; Simakov, O.; Chapman, J.; Fahey, B.; Gauthier, M.E.A.; Mitros, T.; Richards, G.S.; Conaco, C.; Dacre, M.; Hellsten, U.; et al. The Amphimedon queenslandica genome and the evolution of animal complexity. Nature 2010, 466, 720–726. [Google Scholar] [CrossRef]
- Fairclough, S.R.; Chen, Z.; Kramer, E.; Zeng, Q.; Young, S.; Robertson, H.M.; Begovic, E.; Richter, D.J.; Russ, C.; Westbrook, M.J.; et al. Premetazoan genome evolution and the regulation of cell differentiation in the choanoflagellate Salpingoeca rosetta. Genome Biol. 2013, 14, R15. [Google Scholar] [CrossRef]
- Schmucker, D.; Clemens, J.C.; Shu, H.; A Worby, C.; Xiao, J.; Muda, M.; E Dixon, J.; Zipursky, S. Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity. Cell 2000, 101, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Nayak, S.; Goree, J.; Schedl, T.; Meyer, B. fog-2 and the evolution of self-fertile hermaphroditism in Caenorhabditis. PLoS Biol. 2005, 3, e6. [Google Scholar] [CrossRef]
- Florio, M.; Albert, M.; Taverna, E.; Namba, T.; Brandl, H.; Lewitus, E.; Haffner, C.; Sykes, A.; Wong, F.K.; Peters, J.; et al. Human-specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science 2015, 347, 1465–1470. [Google Scholar] [CrossRef]
- Suzuki, I.K.; Gacquer, D.; Van Heurck, R.; Kumar, D.; Wojno, M.; Bilheu, A.; Herpoel, A.; Lambert, N.; Cheron, J.; Polleux, F.; et al. Human-specific NOTCH2NL genes expand cortical neurogenesis through Delta/Notch regulation. Cell 2018, 173, 1370–1384.e16. [Google Scholar] [CrossRef]
- Noh, B.; Amasino, R.M. PIE1, an ISWI family gene, is required for FLC activation and floral repression in Arabidopsis. Plant Cell 2003, 15, 1671–1682. [Google Scholar] [CrossRef] [PubMed]
- Lotan, T.; Ohto, M.-A.; Yee, K.M.; West, M.A.; Lo, R.; Kwong, R.W.; Yamagishi, K.; Fischer, R.L.; Goldberg, R.B.; Harada, J.J. Arabidopsis LEAFY COTYLEDON1 is sufficient to induce embryo development in vegetative cells. Cell 1998, 93, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M. The Origins of Genome Architecture; Sinauer Associates: Sunderland, MA, USA, 2007; ISBN 978-0-87893-484-3. [Google Scholar]
- Schaschl, H.; Wallner, B. Population-specific, recent positive directional selection suggests adaptation of human male reproductive genes to different environmental conditions. BMC Evol. Biol. 2020, 20, 27. [Google Scholar] [CrossRef]
- Chen, J.; He, X.; Jakovlić, I. Positive selection-driven fixation of a hominin-specific amino acid mutation related to dephosphorylation in IRF9. BMC Ecol. Evol. 2022, 22, 132. [Google Scholar] [CrossRef]
- Zhao, L.; Svetec, N.; Begun, D.J. De Novo Genes. Annu. Rev. Genet. 2024, 58, 211–232. [Google Scholar] [CrossRef]
- Artieri, C.G.; Fraser, H.B. Evolution at two levels of gene expression in yeast. Genome Res. 2014, 24, 411–421. [Google Scholar] [CrossRef]
- Long, M.; VanKuren, N.W.; Chen, S.; Vibranovski, M.D. New gene evolution: Little did we know. Annu. Rev. Genet. 2013, 47, 307–333. [Google Scholar] [CrossRef] [PubMed]
- Force, A.; Lynch, M.; Pickett, F.B.; Amores, A.; Yan, Y.L.; Postlethwait, J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 1999, 151, 1531–1545. [Google Scholar] [CrossRef] [PubMed]
- Van de Peer, Y.; Maere, S.; Meyer, A. The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 2009, 10, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Soltis, P.S.; Marchant, D.B.; Van de Peer, Y.; Soltis, D.E. Polyploidy and genome evolution in plants. Curr. Opin. Genet. Dev. 2015, 35, 119–125. [Google Scholar] [CrossRef]
- Wendel, J.F.; Lisch, D.; Hu, G.; Mason, A.S. The long and short of doubling down: Polyploidy, epigenetics, and the temporal dynamics of genome fractionation. Curr. Opin. Genet. Dev. 2018, 49, 1–7. [Google Scholar] [CrossRef]
- Federico, C.; Cantarella, C.D.; Di Mare, P.; Tosi, S.; Saccone, S. The radial arrangement of the human chromosome 7 in the lymphocyte cell nucleus is associated with chromosomal band gene density. Chromosoma 2008, 117, 399–410. [Google Scholar] [CrossRef]
- Federico, C.; Scavo, C.; Cantarella, C.D.; Motta, S.; Saccone, S.; Bernardi, G. Gene-rich and gene-poor chromosomal regions have different locations in the interphase nuclei of cold-blooded vertebrates. Chromosoma 2006, 115, 123–128. [Google Scholar] [CrossRef]
- Federico, C.; Pappalardo, A.M.; Ferrito, V.; Tosi, S.; Saccone, S. Genomic properties of chromosomal bands are linked to evolutionary rearrangements and new centromere formation in primates. Chromosome Res. 2017, 25, 261–276. [Google Scholar] [CrossRef]
- Gulino, G.M.; Bruno, F.; Sturiale, V.; Brancato, D.; Ragusa, D.; Tosi, S.; Saccone, S.; Federico, C. From FISH to Hi-C: The Chromatin Architecture of the Chromosomal Region 7q36.3, Frequently Rearranged in Leukemic Cells, Is Evolutionary Conserved. Int. J. Mol. Sci. 2021, 22, 2338. [Google Scholar] [CrossRef]
- Brancato, D.; Bruno, F.; Coniglio, E.; Sturiale, V.; Saccone, S.; Federico, C. The Chromatin Organization Close to SNP rs12913832, Involved in Eye Color Variation, Is Evolutionary Conserved in Vertebrates. Int. J. Mol. Sci. 2024, 25, 6602. [Google Scholar] [CrossRef]
- Federico, C.; Brancato, D.; Bruno, F.; Galvano, D.; Caruso, M.; Saccone, S. Robertsonian Translocation between Human Chromosomes 21 and 22, Inherited across Three Generations, without Any Phenotypic Effect. Genes 2024, 15, 722. [Google Scholar] [CrossRef] [PubMed]
- Bridger, J.M.; Foeger, N.; Kill, I.R.; Herrmann, H. The nuclear lamina. Both a structural framework and a platform for genome organization. FEBS J. 2007, 274, 1354–1361. [Google Scholar] [CrossRef]
- Szczerbal, I.; Foster, H.A.; Bridger, J.M. The spatial repositioning of adipogenesis genes is correlated with their expression status in a porcine mesenchymal stem cell adipogenesis model system. Chromosoma 2009, 118, 647–663. [Google Scholar] [CrossRef]
- Fraser, P.; Bickmore, W. Nuclear organization of the genome and the potential for gene regulation. Nature 2007, 447, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Lanctôt, C.; Cheutin, T.; Cremer, M.; Cavalli, G.; Cremer, T. Dynamic genome architecture in the nuclear space: Regulation of gene expression in three dimensions. Nat. Rev. Genet. 2007, 8, 104–115. [Google Scholar] [CrossRef]
- Cremer, T.; Cremer, M.; Hübner, B.; Strickfaden, H.; Smeets, D.; Popken, J.; Sterr, M.; Markaki, Y.; Rippe, K.; Cremer, C. The 4D nucleome: Evidence for a dynamic nuclear landscape based on co-aligned active and inactive nuclear compartments. FEBS Lett. 2015, 589, 2931–2943. [Google Scholar] [CrossRef]
- Bickmore, W.A. The spatial organization of the human genome. Annu. Rev. Genom. Hum. Genet. 2013, 14, 67–84. [Google Scholar] [CrossRef]
- Conant, G.C.; Wolfe, K.H. Turning a hobby into a job: How duplicated genes find new functions. Nat. Rev. Genet. 2008, 9, 938–950. [Google Scholar] [CrossRef] [PubMed]
- Carroll, S.B. Evolution at two levels: On genes and form. PLoS Biol. 2005, 3, e245. [Google Scholar] [CrossRef]
- Wray, G.A. The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 2007, 8, 206–216. [Google Scholar] [CrossRef]
- Rodin, S.N.; Riggs, A.D. Epigenetic silencing may aid evolution by gene duplication. J. Mol. Evol. 2003, 56, 718–729. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005, 579, 3346–3354. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, C.J. Moonlighting Proteins: Proteins with Multiple Functions. In Molecular Chaperones and Cell Signalling; Henderson, B., Pockley, A.G., Eds.; Cambridge University Press: Cambridge, UK, 2005; pp. 61–77. [Google Scholar] [CrossRef]
- Niimura, Y. Olfactory receptor multigene family in vertebrates: From the viewpoint of evolutionary genomics. Curr. Genom. 2012, 13, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.P.; Amemiya, C.; Ruddle, F. Hox cluster duplications and the opportunity for evolutionary novelties. Proc. Natl. Acad. Sci. USA 2003, 100, 14603–14606. [Google Scholar] [CrossRef]
- Lemons, D.; McGinnis, W. Genomic evolution of Hox gene clusters. Science 2006, 313, 1918–1922. [Google Scholar] [CrossRef]
- Imbeault, M.; Helleboid, P.Y.; Trono, D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 2017, 543, 550–554. [Google Scholar] [CrossRef]
- Dennis, M.Y.; Nuttle, X.; Sudmant, P.H.; Antonacci, F.; Graves, T.A.; Nefedov, M.; Rosenfeld, J.A.; Sajjadian, S.; Malig, M.; Kotkiewicz, H.; et al. Evolution of human-specific neural SRGAP2 genes by incomplete segmental duplication. Cell 2012, 149, 912–922. [Google Scholar] [CrossRef]
- Hardies, S.C.; Edgell, M.H.; Hutchison, C.A. Evolution of the mammalian beta-globin gene cluster. J. Biol. Chem. 1984, 259, 3748–3756. [Google Scholar] [CrossRef]
- Forget, B.G. Developmental control of human globin gene expression. Prog. Clin. Biol. Res. 1990, 352, 313–322. [Google Scholar]
- Stamatoyannopoulos, G. Control of globin gene expression during development and erythroid differentiation. Exp. Hematol. 2005, 33, 259–271. [Google Scholar] [CrossRef]
- Li, Q.; Peterson, K.R.; Fang, X.; Stamatoyannopoulos, G. Locus control regions. Blood 2002, 100, 3077–3086. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Orkin, S.H. The switch from fetal to adult hemoglobin. Cold Spring Harb. Perspect. Med. 2013, 3, a011643. [Google Scholar] [CrossRef] [PubMed]
- Langdon, S.D.; Kaufman, R.E. Gamma-globin gene promoter elements required for interaction with globin enhancers. Blood 1998, 91, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Menne, T.F.; Xu, J.; Akie, T.E.; Lettre, G.; Van Handel, B.; Mikkola, H.K.A.; Hirschhorn, J.N.; Cantor, A.B.; Orkin, S.H. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 2008, 322, 1839–1842. [Google Scholar] [CrossRef]
- Lefèvre, C.M.; Sharp, J.A.; Nicholas, K.R. Evolution of lactation: Ancient origin and extreme adaptations of the lactation system. Annu. Rev. Genom. Hum. Genet. 2010, 11, 219–238. [Google Scholar] [CrossRef]
- Kawasaki, K.; Weiss, K.M. Mineralized tissue and vertebrate evolution: The secretory calcium-binding phosphoprotein gene cluster. Proc. Natl. Acad. Sci. USA 2003, 100, 4060–4065. [Google Scholar] [CrossRef]
- Kawasaki, K.; Lafont, A.G.; Sire, J.Y. The evolution of milk casein genes from tooth genes before the origin of mammals. Mol. Biol. Evol. 2011, 28, 2053–2061. [Google Scholar] [CrossRef]
- Ruiz-Orera, J.; Hernandez-Rodriguez, J.; Chiva, C.; Sabidó, E.; Kondova, I.; Bontrop, R.; Marqués-Bonet, T.; Albà, M.; Noonan, J. Origins of De Novo Genes in Human and Chimpanzee. PLoS Genet. 2015, 11, e1005721. [Google Scholar] [CrossRef]
- Anderson, D.M.; Anderson, K.M.; Chang, C.L.; Makarewich, C.A.; Nelson, B.R.; McAnally, J.R.; Kasaragod, P.; Shelton, J.M.; Liou, J.; Bassel-Duby, R.; et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 2005, 160, 595–606. [Google Scholar] [CrossRef]
- van Heesch, S.; Witte, F.; Schneider-Lunitz, V.; Schulz, J.F.; Adami, E.; Faber, A.B.; Kirchner, M.; Maatz, H.; Blachut, S.; Sandmann, C.L.; et al. The translational landscape of the human heart. Cell 2019, 178, 242–260.e29. [Google Scholar] [CrossRef]
- Kalebic, N.; Gilardi, C.; Stepien, B.; Wilsch-Bräuninger, M.; Long, K.R.; Namba, T.; Florio, M.; Langen, B.; Lombardot, B.; Shevchenko, A.; et al. Human-specific ARHGAP11B induces hallmarks of neocortical expansion in developing ferret neocortex. eLife 2018, 7, e41241. [Google Scholar] [CrossRef] [PubMed]
- Kanton, S.; Boyle, M.J.; He, Z.; Santel, M.; Weigert, A.; Sanchís-Calleja, F.; Guijarro, P.; Sidow, L.; Fleck, J.S.; Han, D.; et al. Organoid single-cell genomic atlas uncovers human-specific features of brain development. Nature 2019, 574, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Ebler, J.; Ebert, P.; Clarke, W.E.; Rausch, T.; Audano, P.A.; Houwaart, T.; Mao, Y.; Korbel, J.O.; Eichler, E.E.; Zody, M.C.; et al. Pangenome-based genome inference allows efficient and accurate genotyping across multiple individuals. Nat. Genet. 2022, 54, 518–525. [Google Scholar] [CrossRef]
- Liu, S.J.; Nowakowski, T.J.; Pollen, A.A.; Lui, J.H.; Horlbeck, M.A.; Attenello, F.J.; He, D.; Weissman, J.S.; Kriegstein, A.R.; Diaz, A.A.; et al. Single-cell analysis of long non-coding RNAs in the developing human neocortex. Genome Biol. 2016, 17, 67. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef]
- Tynianskaia, L.; Heide, M. Human-specific genetic hallmarks in neocortical development: Focus on neural progenitors. Curr. Opin. Genet. Dev. 2024, 89, 102267. [Google Scholar] [CrossRef]
- McLean, C.Y.; Reno, P.L.; Pollen, A.A.; Bassan, A.I.; Capellini, T.D.; Guenther, C.; Indjeian, V.B.; Lim, X.; Menke, D.B.; Schaar, B.T.; et al. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature 2011, 471, 216–219. [Google Scholar] [CrossRef]
- Eberle, M.A.; Fritzilas, E.; Krusche, P.; Källberg, M.; Moore, B.L.; Bekritsky, M.A.; Iqbal, Z.; Chuang, H.-Y.; Humphray, S.J.; Halpern, A.L.; et al. A reference data set of 5.4 million phased human variants validated by genetic inheritance from sequencing a three-generation 17-member pedigree. Genome Res. 2017, 27, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Chaisson, M.J.P.; Sanders, A.D.; Zhao, X.; Malhotra, A.; Porubsky, D.; Rausch, T.; Gardner, E.J.; Rodriguez, O.L.; Guo, L.; Collins, R.L.; et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019, 10, 1784. [Google Scholar] [CrossRef]
- Liao, W.W.; Asri, M.; Ebler, J.; Doerr, D.; Haukness, M.; Hickey, G.; Lu, S.; Lucas, J.K.; Monlong, J.; Abel, H.J.; et al. A draft human pangenome reference. Nature 2023, 617, 312–324. [Google Scholar] [CrossRef]
| Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).


 ,
, 


{kind=link}
{kind=link}