
Integrated Analysis of miRNA and mRNA Expression Profiles Associated with Development of Skeletal Muscle of Jiangquan Black Pigs
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals
2.3. Paraffin Section and H&E Staining
2.4. RNA Extraction, Library Construction, and RNA Sequencing
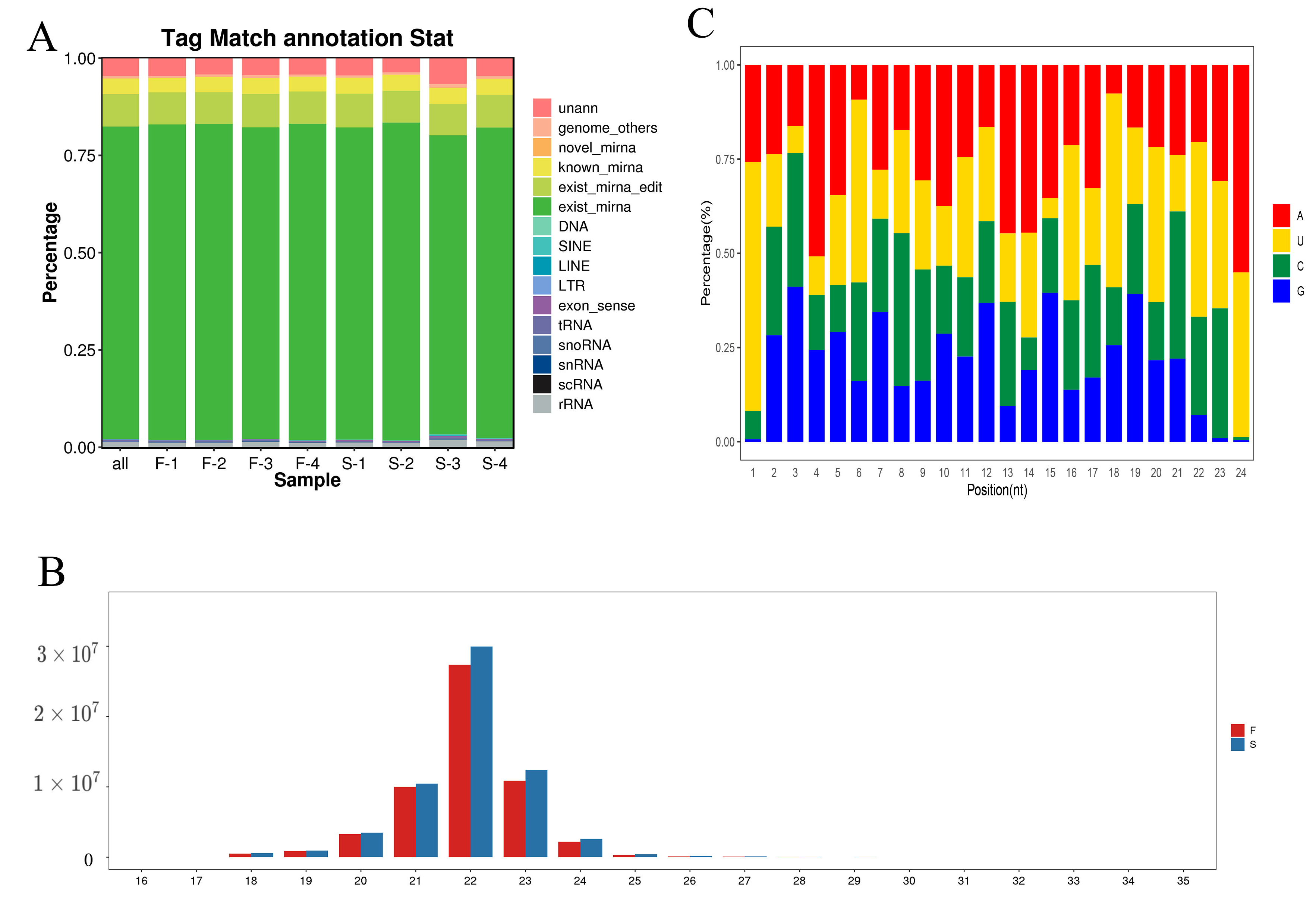
2.5. Mapping and Assembly of Sequenced RNA Data
2.6. Analysis of Differentially Expressed mRNAs and miRNAs
2.7. Functional Enrichment Analysis
2.8. Protein–Protein Interaction (PPI) Network Establishment
2.9. Target Gene Prediction of Differentially Expressed miRNAs
2.10. Quantitative Real-Time Polymerase Chain Reaction Analysis
2.11. Statistical Analysis
3. Results
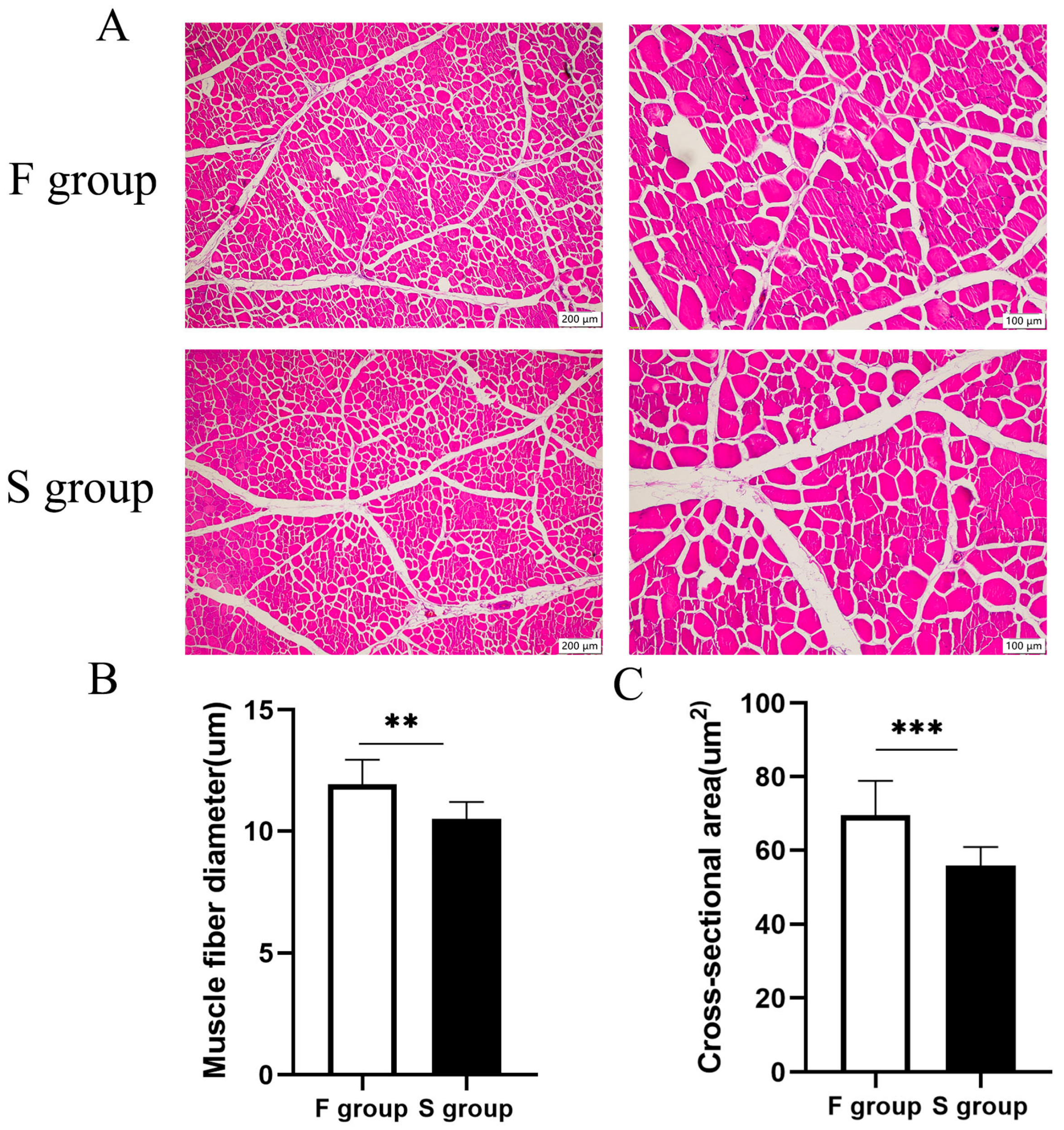
3.1. Comparison of Morphological Characteristics of Muscle Fibers
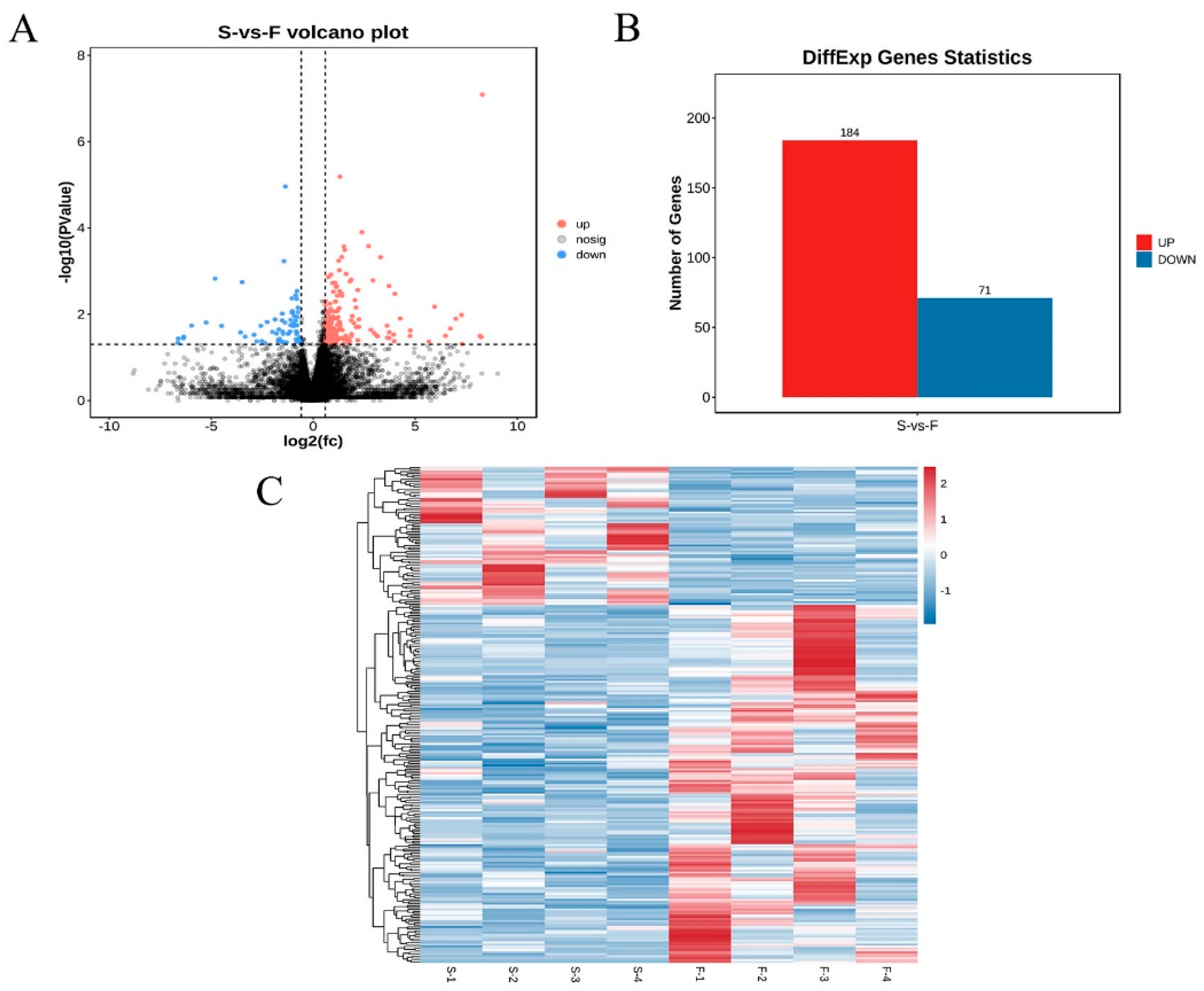
3.2. Identification of Differentially Expressed mRNAs
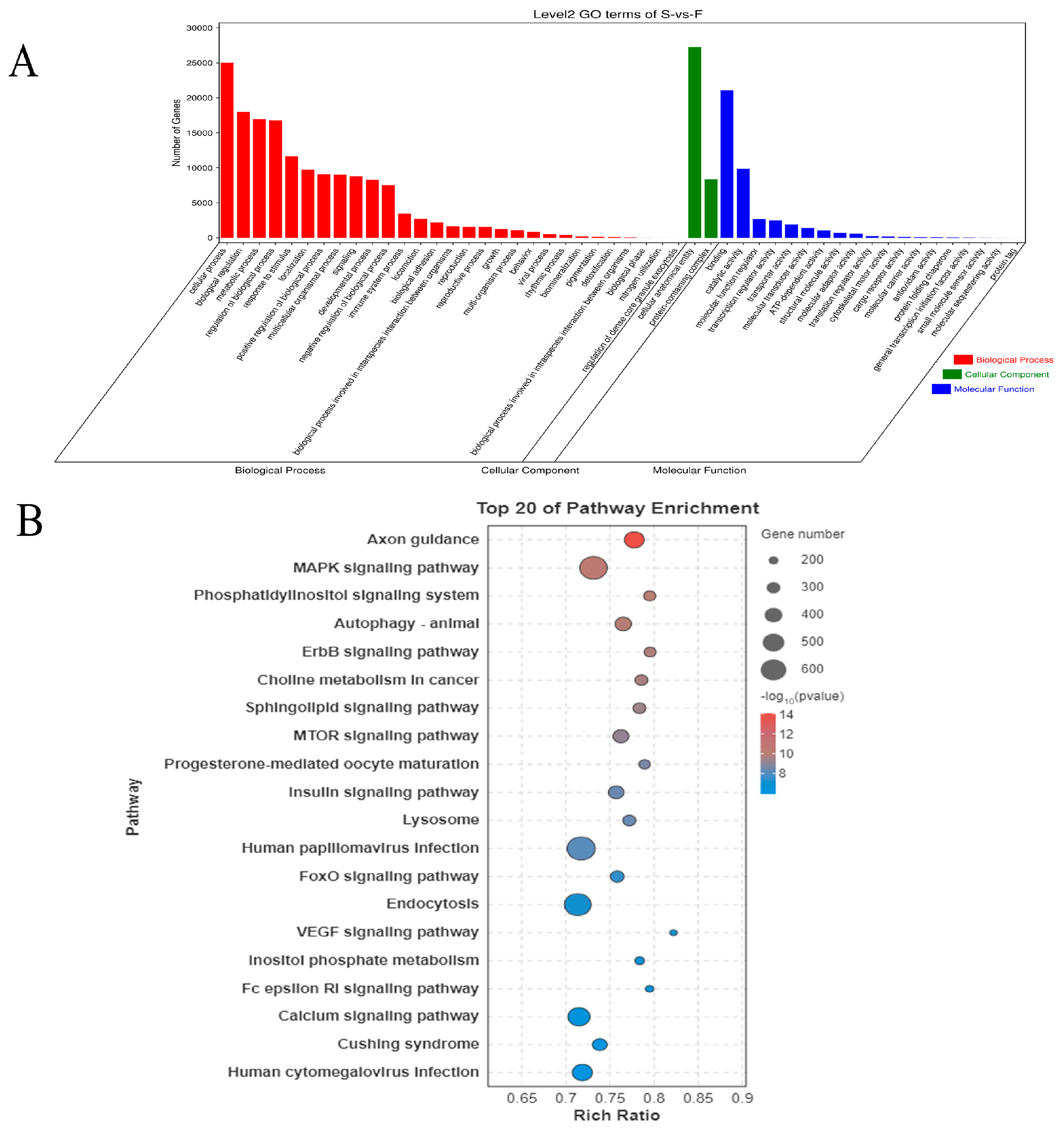
3.3. Functional Enrichment of Differentially Expressed mRNAs
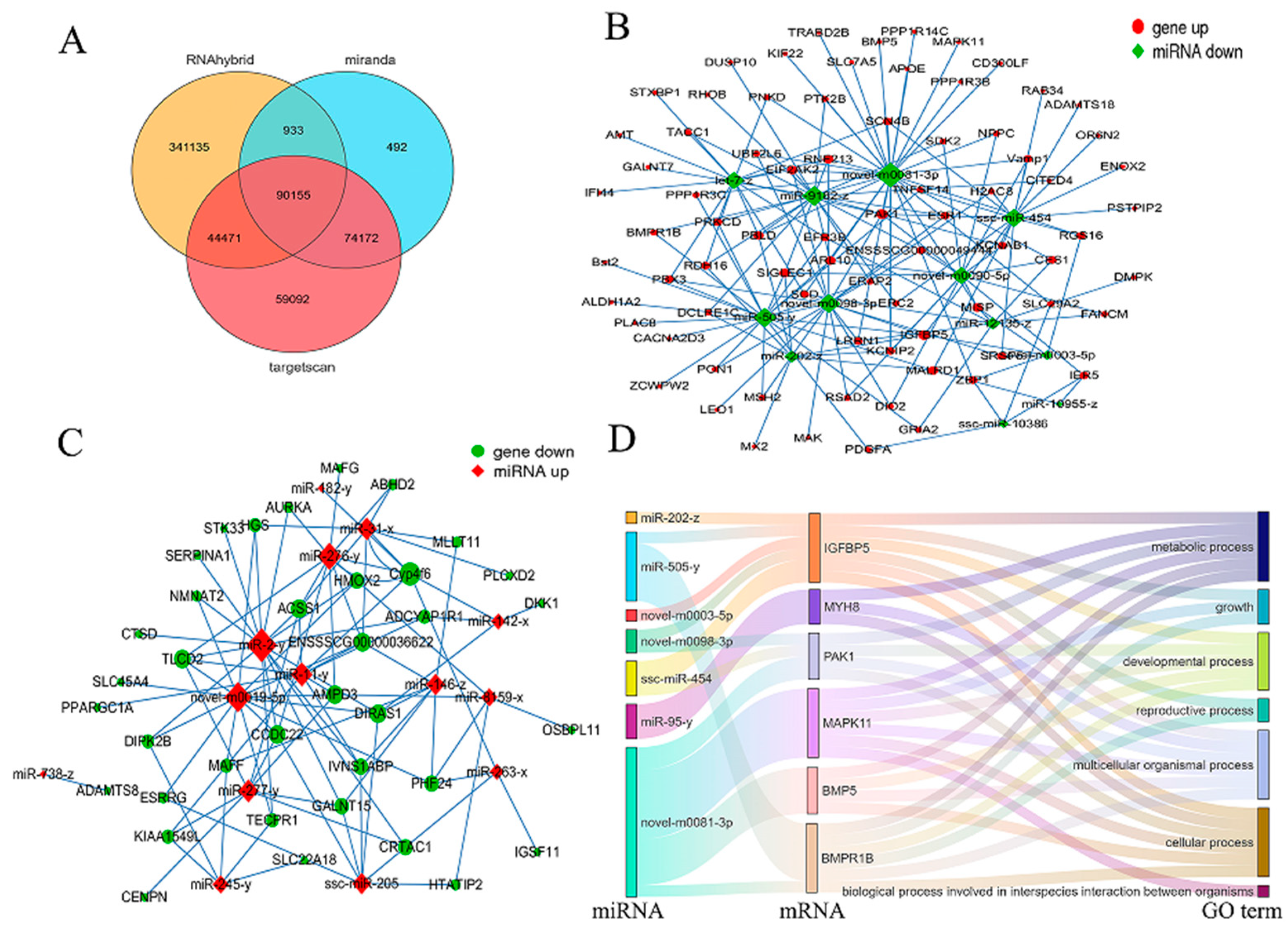
3.4. DEmRNA-Mediated Protein–Protein Interaction Network Analysis
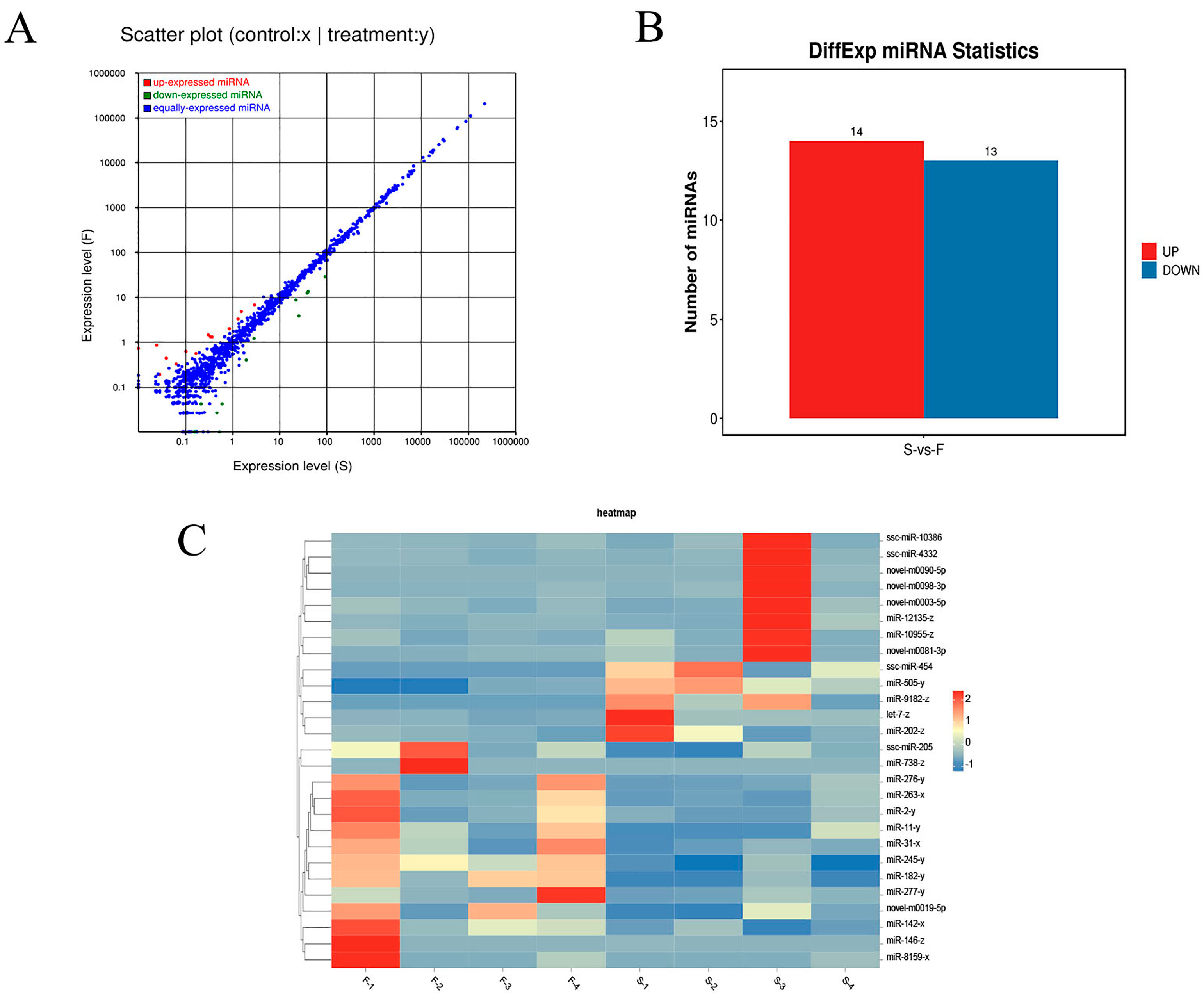
3.5. Identification of Differentially Expressed miRNAs
3.6. Functional Enrichment of Target Genes of Differentially Expressed miRNAs
3.7. Regulatory Network Analysis of DE miRNAs-DEmRNAs
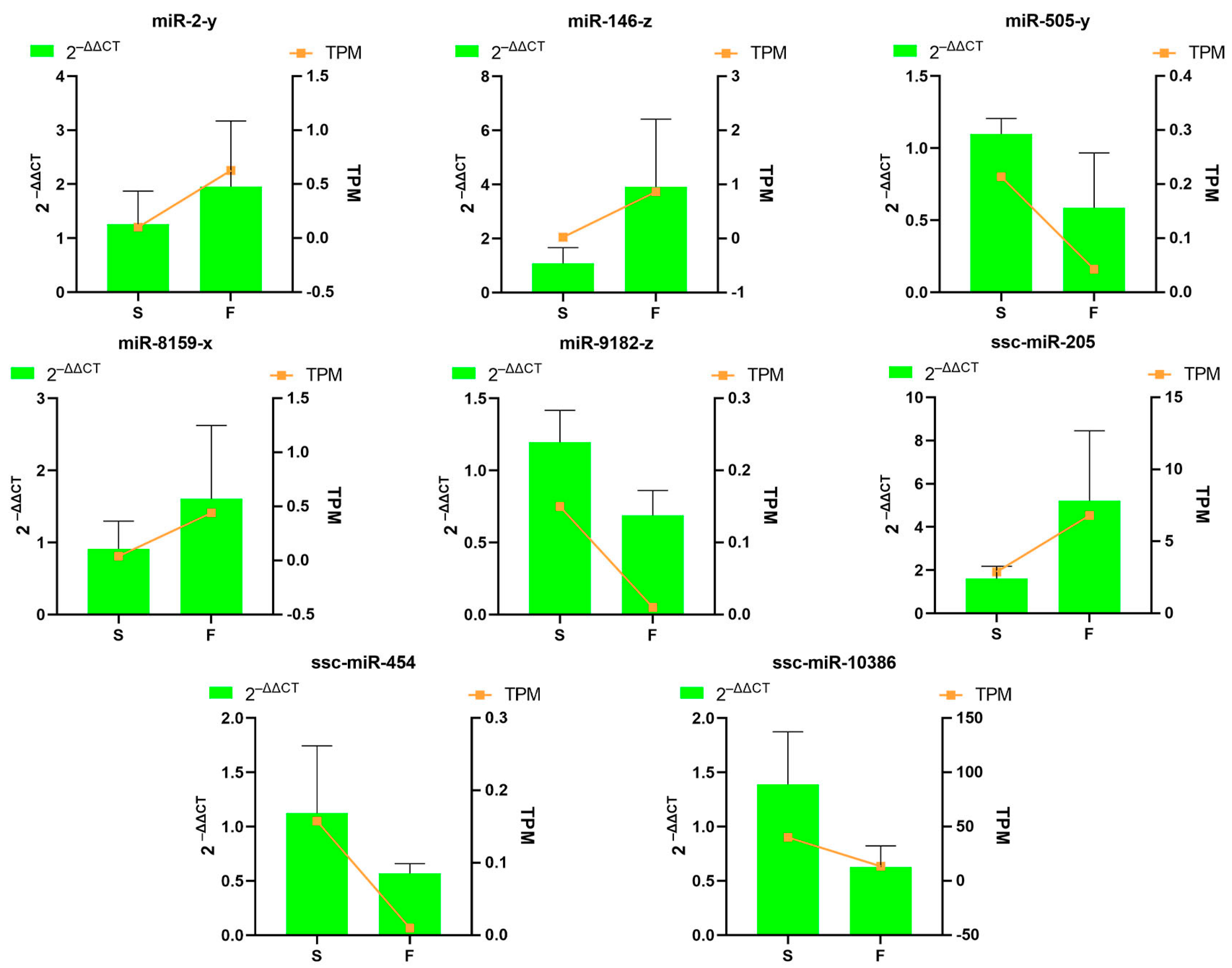
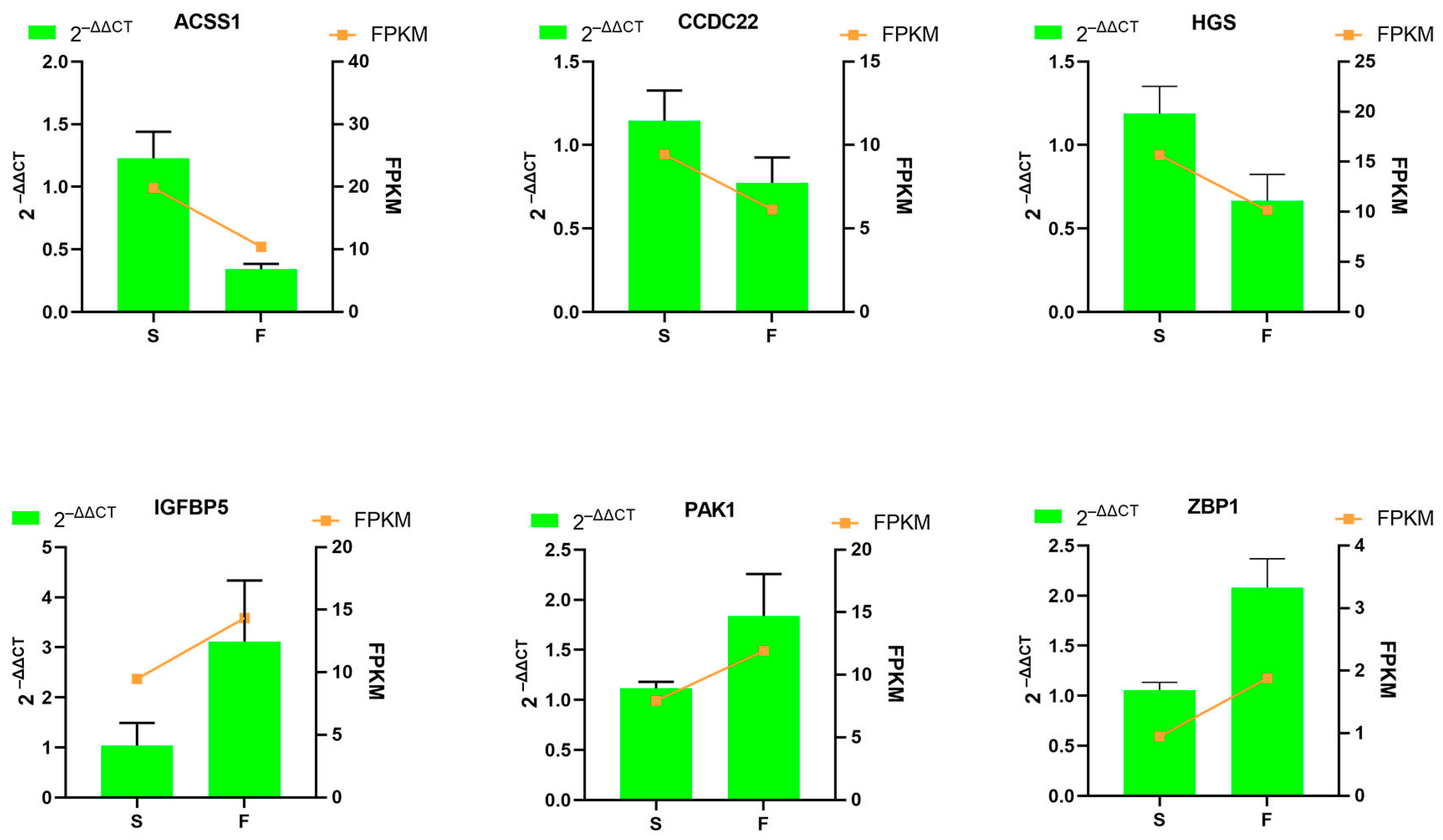
3.8. Validation of Differentially Expressed Genes by Quantitative Real-Time Polymerase Chain Reaction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baskin, K.K.; Winders, B.R.; Olson, E.N. Muscle as a “mediator” of systemic metabolism. Cell Metab. 2015, 21, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Listrat, A.; Lebret, B.; Louveau, I.; Astruc, T.; Bonnet, M.; Lefaucheur, L.; Picard, B.; Bugeon, J. How Muscle Structure and Composition Influence Meat and Flesh Quality. Sci. World J. 2016, 2016, 3182746. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhao, X.; Xue, H.; Zou, X.; Liu, G.; Deng, M.; Sun, B.; Guo, Y.; Liu, D.; Li, Y. RNA-Seq Reveals miRNA and mRNA Co-regulate Muscle Differentiation in Fetal Leizhou Goats. Front. Vet. Sci. 2022, 9, 829769. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Bofill-De Ros, X.; Yang, A.; Gu, S. IsomiRs: Expanding the miRNA repression toolbox beyond the seed. BBA Gene Regul. Mech. 2020, 1863, 194373. [Google Scholar] [CrossRef]
- Sweetman, D.; Goljanek, K.; Rathjen, T.; Oustanina, S.; Braun, T.; Dalmay, T.; Münsterberg, A. Specific requirements of MRFs for the expression of muscle specific microRNAs, miR-1, miR-206 and miR-133. Dev. Biol. 2008, 321, 491–499. [Google Scholar] [CrossRef]
- Sui, M.; Zheng, Q.; Wu, H.; Zhu, L.; Ling, Y.; Wang, L.J.; Fang, F.; Liu, Y.; Zhang, Z.; Chu, M.; et al. The expression and regulation of miR-1 in goat skeletal muscle and satellite cell during muscle growth and development. Anim. Biotechnol. 2020, 31, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chen, Q.; Bao, J.; Pu, Y.; Han, J.; Zhao, H.; Ma, Y.; Zhao, Q. Genome-Wide Analysis of Circular RNAs Reveals circCHRNG Regulates Sheep Myoblast Proliferation via miR-133/SRF and MEF2A Axis. Int. J. Mol. Sci. 2022, 23, 16065. [Google Scholar] [CrossRef]
- Hanna, J.A.; Garcia, M.R.; Go, J.C.; Finkelstein, D.; Kodali, K.; Pagala, V.; Wang, X.; Peng, J.; Hatley, M.E. PAX7 is a required target for microRNA-206-induced differentiation of fusion-negative rhabdomyosarcoma. Cell Death Dis. 2016, 7, e2256. [Google Scholar] [CrossRef]
- Sato, T.; Yamamoto, T.; Sehara-Fujisawa, A. miR-195/497 induce postnatal quiescence of skeletal muscle stem cells. Nat. Commun. 2014, 5, 4597. [Google Scholar] [CrossRef]
- Hou, L.; Xu, J.; Li, H.; Ou, J.; Jiao, Y.; Hu, C.; Wang, C. MiR-34c represses muscle development by forming a regulatory loop with Notch1. Sci. Rep. 2017, 7, 9346. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.H.; Charville, G.W.; Edalati, A.; Quach, N.L.; Park, L.; Yoo, B.; Liu, L.; Rando, T.A.; Hoang, P. Maintenance of muscle stem-cell quiescence by microRNA-489. Nature 2012, 482, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, S.; Qin, M.; Chen, W.; Zeng, Y. Identification and Functional Prediction of Circular RNAs Related to Growth Traits and Skeletal Muscle Development in Duroc pigs. Front. Genet. 2022, 13, 858763. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Qin, M.; Zhang, Y.; Xue, H.; Li, S.; Chen, W.; Zeng, Y. Identification and functional prediction of long non-coding RNAs related to skeletal muscle development in Duroc pigs. Anim. Biosci. 2022, 35, 1512–1523. [Google Scholar] [CrossRef]
- Khatri, P.; Voichita, C.; Kattan, K.; Ansari, N.; Khatri, A.; Georgescu, C.; Tarca, A.L.; Draghici, S. Onto-Tools: New additions and improvements in 2006. Nucleic Acids Res. 2007, 35, W206–W211. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]
- Enright, A.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef]
- Merz, K.E.; Thurmond, D.C. Role of Skeletal Muscle in Insulin Resistance and Glucose Uptake. Compr. Physiol. 2020, 10, 785–809. [Google Scholar] [CrossRef]
- Chen, B.; Yue, Y.; Li, J.; Liu, J.; Yuan, C.; Guo, T.; Zhang, D.; Yang, B.; Lu, Z. Transcriptome-metabolome analysis reveals how sires affect meat quality in hybrid sheep populations. Front. Nutr. 2022, 9, 967985. [Google Scholar] [CrossRef]
- Kong, L.; Yuan, C.; Guo, T.; Sun, L.; Liu, J.; Lu, Z. Inhibitor of Myom3 inhibits proliferation and promotes differentiation of sheep myoblasts. Genomics 2024, 116, 110921. [Google Scholar] [CrossRef]
- Israeli, D.; Poupiot, J.; Amor, F.; Charton, K.; Lostal, W.; Jeanson-Leh, L.; Richard, I. Circulating miRNAs are generic and versatile therapeutic monitoring biomarkers in muscular dystrophies. Sci. Rep. 2016, 6, 28097. [Google Scholar] [CrossRef]
- Song, Q.; Li, J.; Li, S.; Cao, H.; Jin, X.; Zeng, Y.; Chen, W. Full-Length Transcriptome Analysis of Skeletal Muscle of Jiangquan Black Pig at Different Developmental Stages. Int. J. Mol. Sci. 2024, 25, 6095. [Google Scholar] [CrossRef] [PubMed]
- Pöllänen, E.; Fey, V.; Törmäkangas, T.; Ronkainen, P.H.A.; Taaffe, D.R.; Takala, T.; Koskinen, S.; Cheng, S.; Puolakka, J.; Kujala, U.M.; et al. Power training and postmenopausal hormone therapy affect transcriptional control of specific co-regulated gene clusters in skeletal muscle. AGE 2010, 32, 347–363. [Google Scholar] [CrossRef]
- Mukherjee, A.; Wilson, E.M.; Rotwein, P. Insulin-like growth factor (igf) binding protein-5 blocks skeletal muscle differentiation by inhibiting IGF actions. Mol. Endocrinol. 2008, 22, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Ren, H.; Gao, S. Insulin-like growth factors (IGFs), IGF receptors, and IGF-binding proteins: Roles in skeletal muscle growth and differentiation. Gen. Comp. Endocrinol. 2010, 167, 344–351. [Google Scholar] [CrossRef]
- James, P.L.; Stewart, C.E.; Rotwein, P. Insulin-like growth factor binding protein-5 modulates muscle differentiation through an insulin-like growth factor-dependent mechanism. J. Cell Biol. 1996, 133, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Barbé, C.; Loumaye, A.; Lause, P.; Ritvos, O.; Thissen, J.P. p21-Activated Kinase 1 Is Permissive for the Skeletal Muscle Hypertrophy Induced by Myostatin Inhibition. Front. Physiol. 2021, 12, 677746. [Google Scholar] [CrossRef]
- Wang, S.; Shi, M.; Zhang, Y.; Niu, J.; Li, W.; Yuan, J.; Cai, C.; Yang, Y.; Gao, P.; Guo, X.; et al. Construction of LncRNA-Related ceRNA Networks in Longissimus Dorsi Muscle of Jinfen White Pigs at Different Developmental Stages. Curr. Issues Mol. Biol. 2024, 46, 340–354. [Google Scholar] [CrossRef]
- Zhao, W.; Li, Z.; Liu, Q.; Xie, S.; Li, M.; Wang, Y.; Li, C. Analysis of long intergenic non-coding RNAs transcriptomic profiling in skeletal muscle growth during porcine embryonic development. Sci. Rep. 2021, 11, 15240. [Google Scholar] [CrossRef]
- Lee, S. Quadrupling muscle mass in mice by targeting TGF-beta signaling pathways. PLoS ONE 2007, 2, e789. [Google Scholar] [CrossRef]
- Dreher, S.I.; Höckele, S.; Huypens, P.; Irmler, M.; Hoffmann, C.; Jeske, T.; Hastreiter, M.; Moller, A.; Birkenfeld, A.L.; Häring, H.-U.; et al. TGF-beta Induction of miR-143/145 Is Associated to Exercise Response by Influencing Differentiation and Insulin Signaling Molecules in Human Skeletal Muscle. Cells 2021, 10, 3443. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Raza, S.H.A.; Yang, Z.; Wang, S.; Yu, S.; Aloufi, B.H.; Li, B.; Zan, L. Bta-miR-181d and Bta-miR-196a mediated proliferation, differentiation, and apoptosis in Bovine Myogenic Cells. J. Anim. Sci. 2024, 102, skae142. [Google Scholar]
- Anderson, C.; Catoe, H.; Werner, R. MIR-206 regulates connexin43 expression during skeletal muscle development. Nucleic Acids Res. 2006, 34, 5863–5871. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, J.J. MicroRNA-206: The skeletal muscle-specific myomiR. Biochim. Biophys. Acta (BBA)-Bioenerg. 2008, 1779, 682–691. [Google Scholar] [CrossRef]
- Torma, F.; Gombos, Z.; Fridvalszki, M.; Langmar, G.; Tarcza, Z.; Merkely, B.; Naito, H.; Ichinoseki-Sekine, N.; Takeda, M.; Murlasits, Z.; et al. Blood flow restriction in human skeletal muscle during rest periods after high-load resistance training down-regulates miR-206 and induces Pax7. J. Sport Health Sci. 2021, 10, 470–477. [Google Scholar] [CrossRef]
- Zhang, X.; Cai, S.; Chen, L.; Yuan, R.; Nie, Y.; Ding, S.; Fang, Y.; Zhu, Q.; Chen, K.; Wei, H.; et al. Integrated miRNA–mRNA transcriptomic analysis reveals epigenetic-mediated embryonic muscle growth differences between Wuzhishan and Landrace pigs. J. Anim. Sci. 2019, 97, 1967–1978. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ma, Y.; Zhao, T.; Li, M.; Mao, Y.; Yang, Z. Epigenetic Regulation Mechanisms of the Cofilin-1 Gene in the Development and Differentiation of Bovine Primary Myoblasts. Genes 2022, 13, 723. [Google Scholar] [CrossRef]
- Ma, J.; Zhu, Y.; Zhou, X.; Zhang, J.; Sun, J.; Li, Z.; Jin, L.; Long, K.; Lu, L.; Ge, L. miR-205 Regulates the Fusion of Porcine Myoblast by Targeting the Myomaker Gene. Cells 2023, 12, 1107. [Google Scholar] [CrossRef]
- Li, Z.; Abdalla, B.A.; Zheng, M.; He, X.; Cai, B.; Han, P.; Ouyang, H.; Chen, B.; Nie, Q.; Zhang, X. Systematic transcriptome-wide analysis of mRNA–miRNA interactions reveals the involvement of miR-142-5p and its target (FOXO3) in skeletal muscle growth in chickens. Mol. Genet. Genom. 2018, 293, 69–80. [Google Scholar] [CrossRef]
- Wang, K.; Liufu, S.; Yu, Z.; Xu, X.; Ai, N.; Li, X.; Liu, X.; Chen, B.; Zhang, Y.; Ma, H.; et al. miR-100-5p Regulates Skeletal Muscle Myogenesis through the Trib2/mTOR/S6K Signaling Pathway. Int. J. Mol. Sci. 2023, 24, 8906. [Google Scholar] [CrossRef]
- Yin, H.; Zhao, J.; He, H.; Chen, Y.; Wang, Y.; Li, D.; Zhu, Q. Gga-miR-3525 Targets PDLIM3 Through the MAPK Signaling Pathway to Regulate the Proliferation and Differentiation of Skeletal Muscle Satellite Cells. Int. J. Mol. Sci. 2020, 21, 5573. [Google Scholar] [CrossRef] [PubMed]
- Kamanga-Sollo, E.; Pampusch, M.; White, M.; Hathaway, M.; Dayton, W. Insulin-like growth factor binding protein (IGFBP)-3 and IGFBP-5 mediate TGF-β- and myostatin-induced suppression of proliferation in porcine embryonic myogenic cell cultures. Exp. Cell Res. 2005, 311, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Dayton, W.R.; White, M.E. Cellular and molecular regulation of muscle growth and development in meat animals1,2. J. Anim. Sci. 2008, 86, E217–E225. [Google Scholar] [CrossRef] [PubMed]
- Berton, M.P.; Fonseca, L.F.S.; Gimenez, D.F.J.; Utembergue, B.L.; Cesar, A.S.M.; Coutinho, L.L.; de Lemos, M.V.A.; Aboujaoude, C.; Pereira, A.S.C.; de O Silva, R.M.; et al. Gene expression profile of intramuscular muscle in Nellore cattle with extreme values of fatty acid. BMC Genom. 2016, 17, 972. [Google Scholar] [CrossRef]
- Xu, G.; Schell, J.; Quan, S.; Gao, Y.; Wei, S.J.; Pan, M.; Han, X.; Li, G.; Zhou, D.; Jiang, H.; et al. Mitochondrial ACSS1-K635 acetylation knock-in mice exhibit altered liver lipid metabolism on a ketogenic diet. Free Radic. Biol. Med. 2025, 232, 260–268. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Clean Data (bp) | Q20 (%) | Q30 (%) | N (%) | GC (%) |
|---|---|---|---|---|---|
| F-1 | 14,169,221,396 | 13,820,127,751 (97.54%) | 13,247,777,202 (93.50%) | 19,211 (0.00%) | 7,920,814,869 (55.90%) |
| F-2 | 15,455,529,768 | 15,078,387,256 (97.56%) | 14,478,846,918 (93.68%) | 194,491 (0.00%) | 8,879,556,177 (57.45%) |
| F-3 | 14,920,893,046 | 14,571,191,202 (97.66%) | 14,003,985,159 (93.85%) | 18,745 (0.00%) | 8,647,668,938 (57.96%) |
| F-4 | 15,350,988,150 | 15,024,061,510 (97.87%) | 14,489,584,711 (94.39%) | 193,383 (0.00%) | 8,869,412,762 (57.78%) |
| S-1 | 15,442,610,918 | 15,051,361,427 (97.47%) | 14,432,699,483 (93.46%) | 195,029 (0.00%) | 8,692,984,968 (56.29%) |
| S-2 | 14,437,471,550 | 14,082,357,667 (97.54%) | 13,524,312,905 (93.68%) | 181,158 (0.00%) | 8,391,017,741 (58.12%) |
| S-3 | 14,095,330,254 | 13,752,865,783 (97.57%) | 13,201,990,565 (93.66%) | 175,519 (0.00%) | 7,982,128,285 (56.63%) |
| S-4 | 14,017,378,016 | 13,671,090,868 (97.53%) | 13,125,187,571 (93.64%) | 176,379 (0.00%) | 8,068,776,013 (57.56%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Li, S.; Chen, W.; Zhang, J.; Ren, Z.; Ma, Z.; Wang, Y.; Zeng, Y. Integrated Analysis of miRNA and mRNA Expression Profiles Associated with Development of Skeletal Muscle of Jiangquan Black Pigs. Genes 2025, 16, 701. https://doi.org/10.3390/genes16060701
Gao Y, Li S, Chen W, Zhang J, Ren Z, Ma Z, Wang Y, Zeng Y. Integrated Analysis of miRNA and mRNA Expression Profiles Associated with Development of Skeletal Muscle of Jiangquan Black Pigs. Genes. 2025; 16(6):701. https://doi.org/10.3390/genes16060701
Chicago/Turabian StyleGao, Yarui, Shiyin Li, Wei Chen, Jianmin Zhang, Zhanchi Ren, Zhao Ma, Yunzhou Wang, and Yongqing Zeng. 2025. "Integrated Analysis of miRNA and mRNA Expression Profiles Associated with Development of Skeletal Muscle of Jiangquan Black Pigs" Genes 16, no. 6: 701. https://doi.org/10.3390/genes16060701
APA StyleGao, Y., Li, S., Chen, W., Zhang, J., Ren, Z., Ma, Z., Wang, Y., & Zeng, Y. (2025). Integrated Analysis of miRNA and mRNA Expression Profiles Associated with Development of Skeletal Muscle of Jiangquan Black Pigs. Genes, 16(6), 701. https://doi.org/10.3390/genes16060701