
Identifying CDCA4 as a Radiotherapy Resistance-Associated Gene in Colorectal Cancer by an Integrated Bioinformatics Analysis Approach

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. WES and Data Processing
2.3. Independent Cohort Data Validation
2.4. scRNA-Seq Data Analysis
2.5. Screening Genes with Differential Expression
2.6. Functional Enrichment Analysis
2.7. Protein–Protein Interaction Network
2.8. Genomic Heterogeneity Analysis
2.9. Statistical Analysis
3. Results
3.1. Mutational Landscape of Resistance to Radiotherapy in CRC
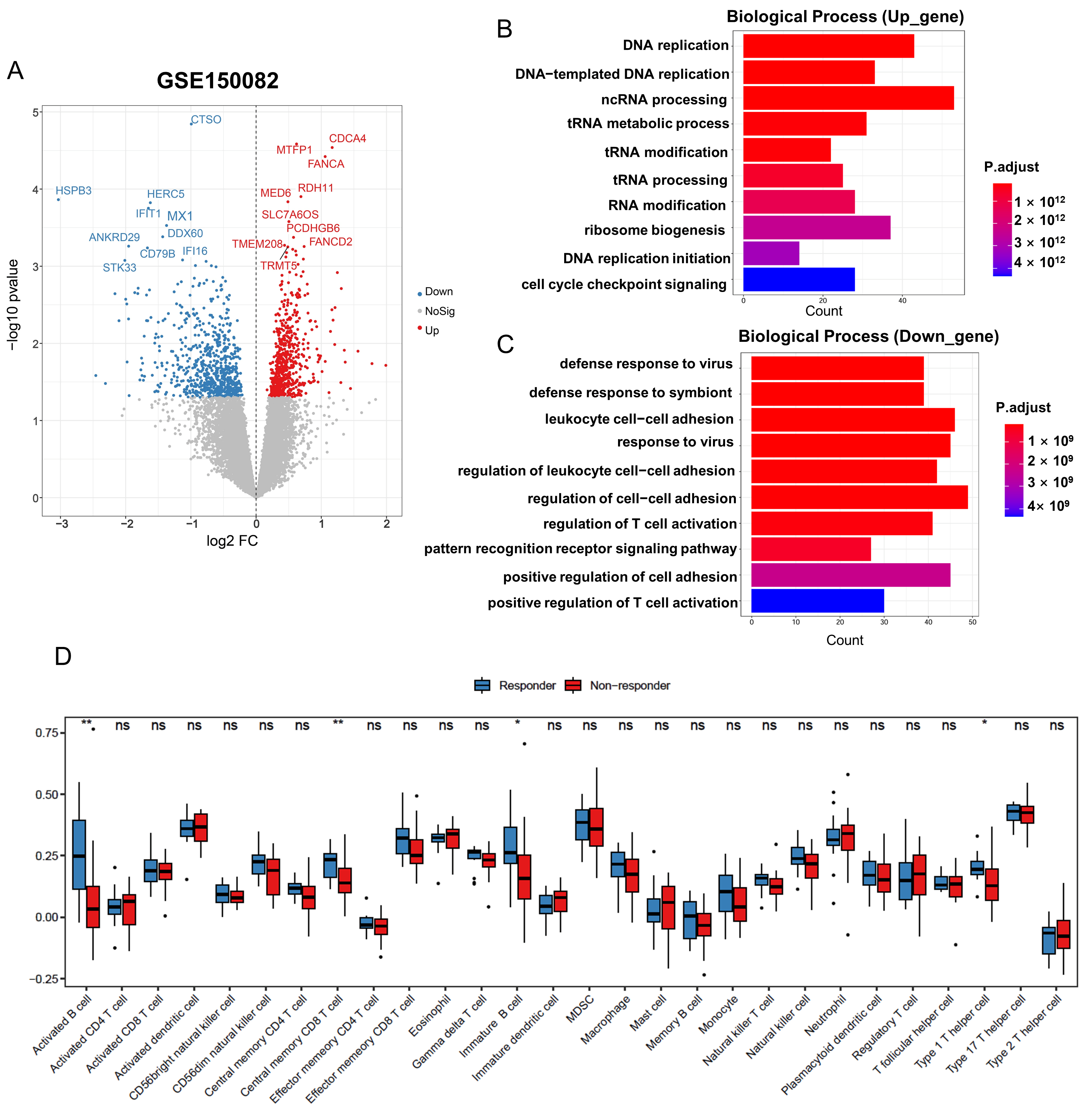
3.2. Identification and Functional Enrichment of DEGs in Radiation-Resistant Rectal Cancer Cohort
3.3. Development of a Gene Signature for Predicting Radiotherapy Efficacy
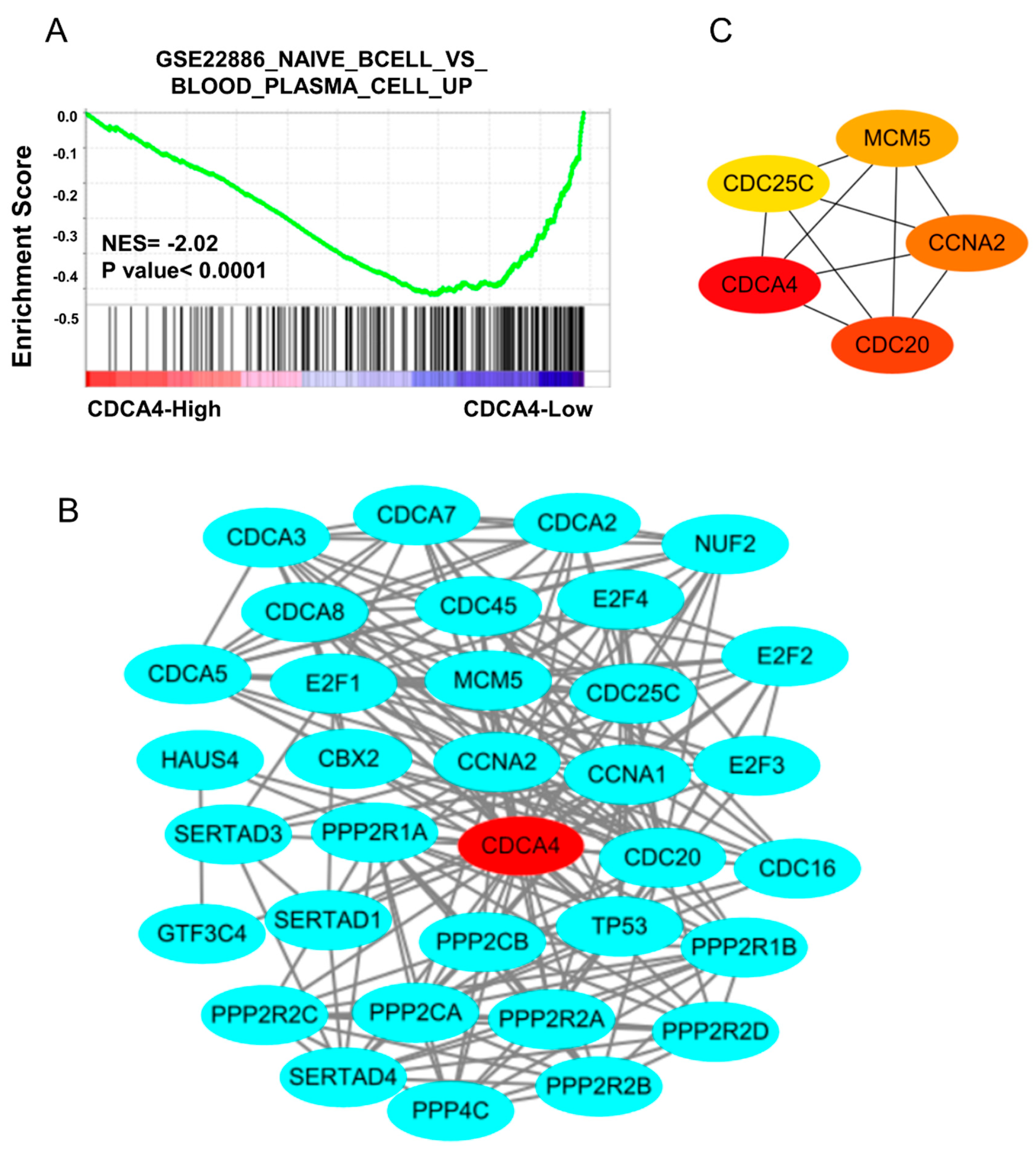
3.4. The Regulatory Network of CDCA4 Affects B Cell Development
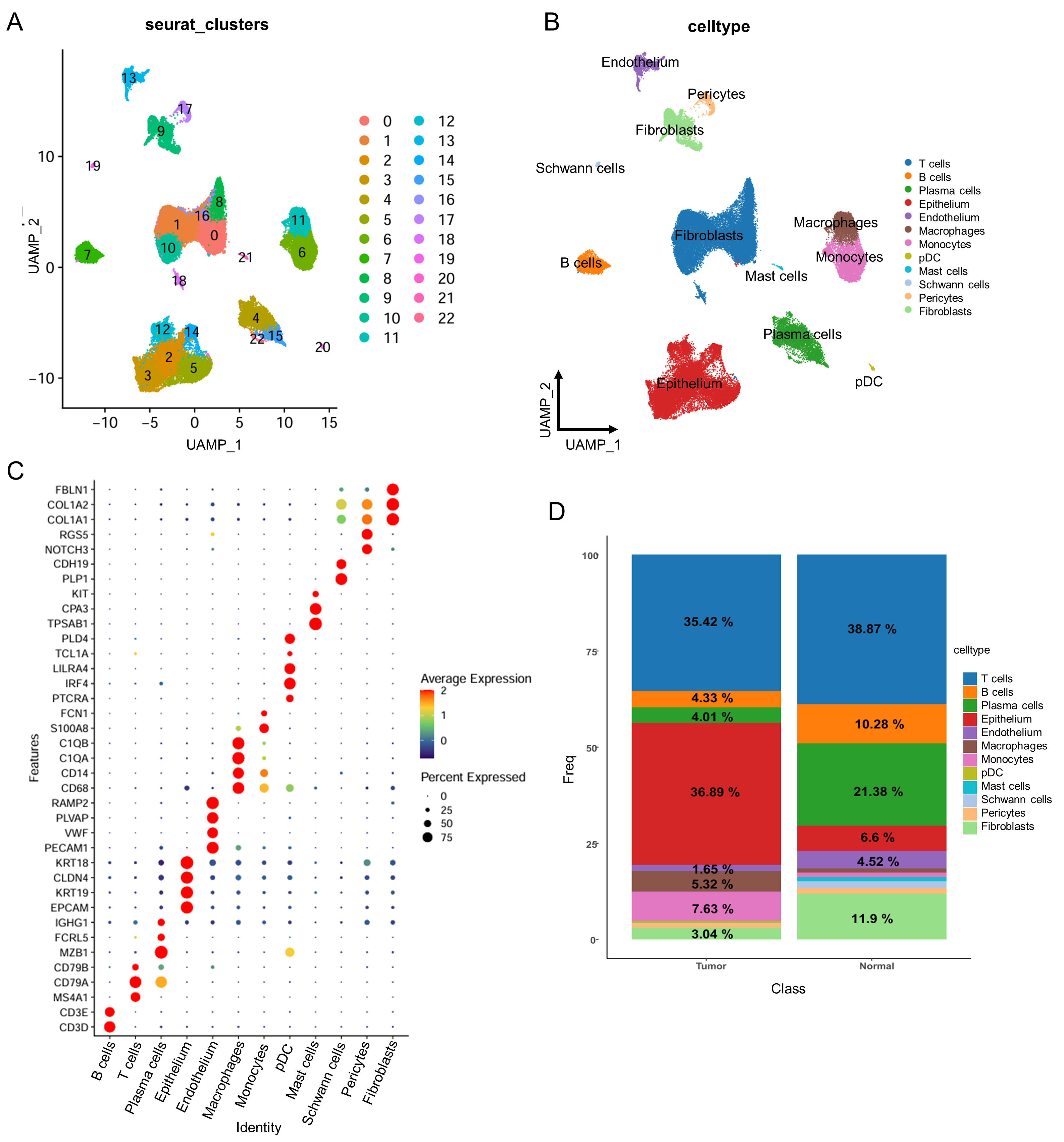
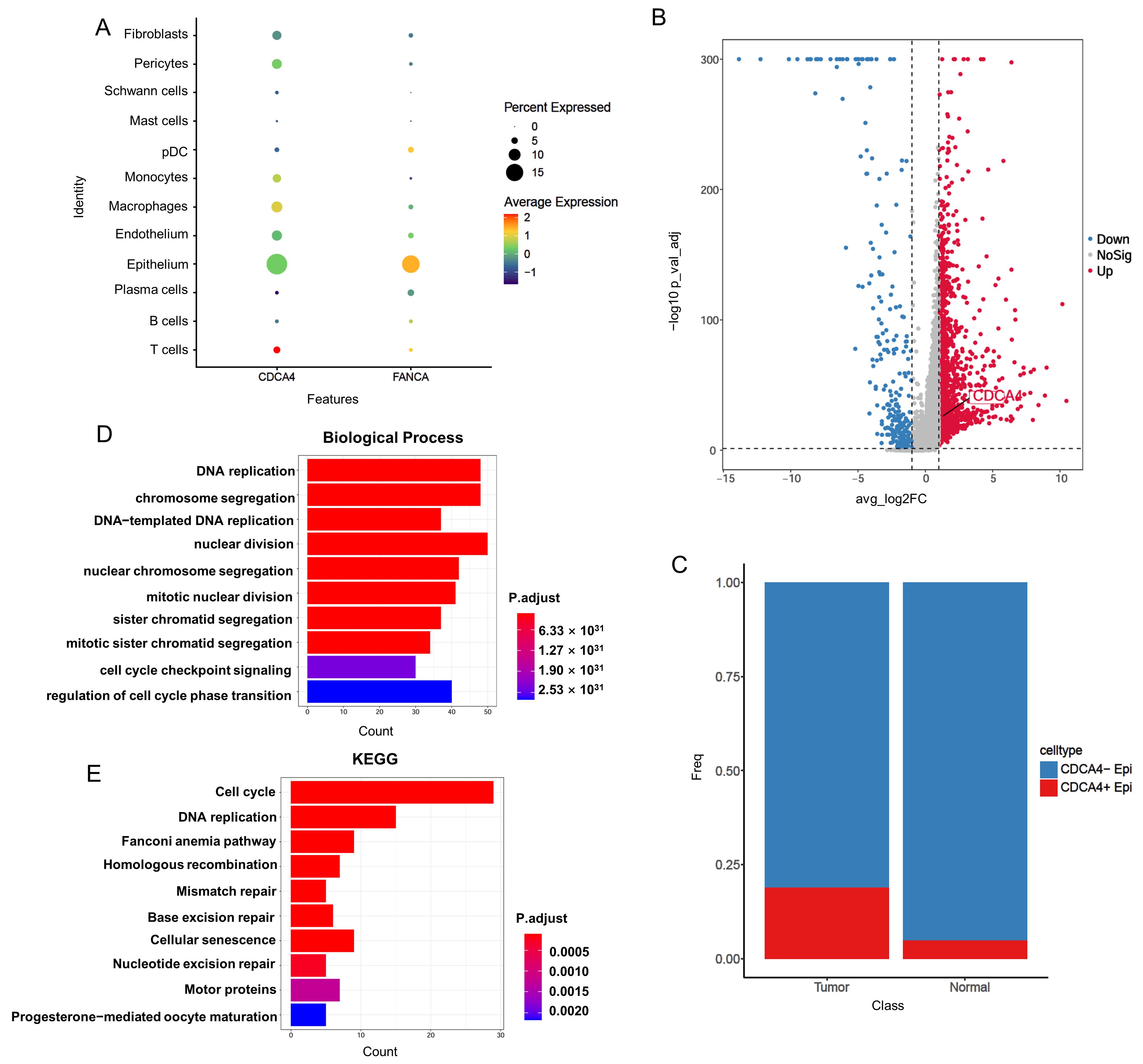
3.5. Single-Cell RNA-Seq Reveals CDCA4 Expression in Epithelial Cells Correlates with Tumor Progression and DNA Repair Pathways
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CRC | Colorectal cancer |
| LARC | Locally advanced rectal cancer |
| pCR | Pathologic complete response |
| nCRT | Neoadjuvant chemoradiotherapy |
| WES | Whole-exome sequencing |
| scRNA-seq | Single-cell RNA sequencing |
| TIME | Tumor immune microenvironment |
| TMB | Tumor mutation burden |
| MSI | Microsatellite instability |
| GWASs | Genome-wide association studies |
| TME | Tumor microenvironment |
| TCGA-READ | The Cancer Genome Atlas–Rectum adenocarcinoma |
| NGS | Next-generation sequencing |
| SNV | Single nucleotide variant |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GSEA | Gene Set Enrichment Analysis |
| ssGSEA | Single-sample GSEA |
| SNPs | Single nucleotide polymorphisms |
| GSVA | Gene Set Variation Analysis |
| ROC | Receiver operating characteristic |
| AUC | Area under the curve |
| TIL-Bs | Tumor-infiltrating B lymphocytes |
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Kamran, S.C.; Lennerz, J.K.; Margolis, C.A.; Liu, D.; Reardon, B.; Wankowicz, S.A.; Van Seventer, E.E.; Tracy, A.; Wo, J.Y.; Carter, S.L.; et al. Integrative Molecular Characterization of Resistance to Neoadjuvant Chemoradiation in Rectal Cancer. Clin. Cancer Res. 2019, 25, 5561–5571. [Google Scholar] [CrossRef]
- Brown, J.M.; Carlson, D.J.; Brenner, D.J. The tumor radiobiology of SRS and SBRT: Are more than the 5 Rs involved? Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 254–262. [Google Scholar] [CrossRef]
- Roh, M.S.; Colangelo, L.H.; O’Connell, M.J.; Yothers, G.; Deutsch, M.; Allegra, C.J.; Kahlenberg, M.S.; Baez-Diaz, L.; Ursiny, C.S.; Petrelli, N.J.; et al. Preoperative multimodality therapy improves disease-free survival in patients with carcinoma of the rectum: NSABP R-03. J. Clin. Oncol. 2009, 27, 5124–5130. [Google Scholar] [CrossRef]
- Minsky, B.D.; Cohen, A.M.; Kemeny, N.; Enker, W.E.; Kelsen, D.P.; Reichman, B.; Saltz, L.; Sigurdson, E.R.; Frankel, J. Enhancement of radiation-induced downstaging of rectal cancer by fluorouracil and high-dose leucovorin chemotherapy. J. Clin. Oncol. 1992, 10, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Thariat, J.; Chevalier, F.; Orbach, D.; Ollivier, L.; Marcy, P.Y.; Corradini, N.; Beddok, A.; Foray, N.; Bougeard, G. Avoidance or adaptation of radiotherapy in patients with cancer with Li-Fraumeni and heritable TP53-related cancer syndromes. Lancet Oncol. 2021, 22, e562–e574. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.S.; Watt, G.P.; Malone, K.E.; Lynch, C.F.; John, E.M.; Knight, J.A.; Woods, M.; Liang, X.; Tischkowitz, M.; Conti, D.V.; et al. Breast Cancer Susceptibility Gene Sequence Variations and Development of Contralateral Breast Cancer. JAMA Netw. Open 2024, 7, e2452158. [Google Scholar] [CrossRef]
- Petry, V.; Bonadio, R.C.; Moutinho, K.; Leite, L.S.; Testa, L.; Cohn, D.; Cagnacci, A.C.; Kim, V.E.H.; Del Pilar Estevez-Diz, M.; Fragoso, M. Frequency of Radiation Therapy-Induced Malignancies in Patients With Li-Fraumeni Syndrome and Early-Stage Breast Cancer and the Influence of Radiation Therapy Technique. Int. J. Radiat. Oncol. Biol. Phys. 2024, 119, 1086–1091. [Google Scholar] [CrossRef]
- Namba, S.; Saito, Y.; Kogure, Y.; Masuda, T.; Bondy, M.L.; Gharahkhani, P.; Gockel, I.; Heider, D.; Hillmer, A.; Jankowski, J.; et al. Common Germline Risk Variants Impact Somatic Alterations and Clinical Features across Cancers. Cancer Res. 2023, 83, 20–27. [Google Scholar] [CrossRef]
- Qian, L.; Lai, X.; Gu, B.; Sun, X. An Immune-Related Gene Signature for Predicting Neoadjuvant Chemoradiotherapy Efficacy in Rectal Carcinoma. Front. Immunol. 2022, 13, 784479. [Google Scholar] [CrossRef]
- Taube, J.M.; Galon, J.; Sholl, L.M.; Rodig, S.J.; Cottrell, T.R.; Giraldo, N.A.; Baras, A.S.; Patel, S.S.; Anders, R.A.; Rimm, D.L.; et al. Implications of the tumor immune microenvironment for staging and therapeutics. Mod. Pathol. 2018, 31, 214–234. [Google Scholar] [CrossRef] [PubMed]
- Mlecnik, B.; Bindea, G.; Angell, H.K.; Maby, P.; Angelova, M.; Tougeron, D.; Church, S.E.; Lafontaine, L.; Fischer, M.; Fredriksen, T.; et al. Integrative Analyses of Colorectal Cancer Show Immunoscore Is a Stronger Predictor of Patient Survival Than Microsatellite Instability. Immunity 2016, 44, 698–711. [Google Scholar] [CrossRef]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pages, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.; Pitroda, S.P.; Weichselbaum, R.R. Radiotherapy, immunity, and immune checkpoint inhibitors. Lancet Oncol. 2024, 25, e352–e362. [Google Scholar] [CrossRef] [PubMed]
- Alganmi, N.; Abusamra, H. Evaluation of an optimized germline exomes pipeline using BWA-MEM2 and Dragen-GATK tools. PLoS ONE 2023, 18, e0288371. [Google Scholar] [CrossRef]
- Hintzsche, J.D.; Robinson, W.A.; Tan, A.C. A Survey of Computational Tools to Analyze and Interpret Whole Exome Sequencing Data. Int. J. Genom. 2016, 2016, 7983236. [Google Scholar] [CrossRef]
- Lin, Y.L.; Chang, P.C.; Hsu, C.; Hung, M.Z.; Chien, Y.H.; Hwu, W.L.; Lai, F.; Lee, N.C. Comparison of GATK and DeepVariant by trio sequencing. Sci. Rep. 2022, 12, 1809. [Google Scholar] [CrossRef]
- Nicolas, A.M.; Pesic, M.; Engel, E.; Ziegler, P.K.; Diefenhardt, M.; Kennel, K.B.; Buettner, F.; Conche, C.; Petrocelli, V.; Elwakeel, E.; et al. Inflammatory fibroblasts mediate resistance to neoadjuvant therapy in rectal cancer. Cancer Cell 2022, 40, 168–184.e13. [Google Scholar] [CrossRef]
- Lee, H.O.; Hong, Y.; Etlioglu, H.E.; Cho, Y.B.; Pomella, V.; Van den Bosch, B.; Vanhecke, J.; Verbandt, S.; Hong, H.; Min, J.W.; et al. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat. Genet. 2020, 52, 594–603. [Google Scholar] [CrossRef]
- Zheng, G.X.; Terry, J.M.; Belgrader, P.; Ryvkin, P.; Bent, Z.W.; Wilson, R.; Ziraldo, S.B.; Wheeler, T.D.; McDermott, G.P.; Zhu, J.; et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017, 8, 14049. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902 e1821. [Google Scholar] [CrossRef] [PubMed]
- Dann, E.; Henderson, N.C.; Teichmann, S.A.; Morgan, M.D.; Marioni, J.C. Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat. Biotechnol. 2022, 40, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Sendoya, J.M.; Iseas, S.; Coraglio, M.; Golubicki, M.; Robbio, J.; Salanova, R.; Kujaruk, M.; Mikolaitis, V.; Rizzolo, M.; Ruiz, G.; et al. Pre-Existing Tumoral B Cell Infiltration and Impaired Genome Maintenance Correlate with Response to Chemoradiotherapy in Locally Advanced Rectal Cancer. Cancers 2020, 12, 2227. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Peng, Z.; Ye, M.; Ding, H.; Feng, Z.; Hu, K. Spatial transcriptomics atlas reveals the crosstalk between cancer-associated fibroblasts and tumor microenvironment components in colorectal cancer. J. Transl. Med. 2022, 20, 302. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.Z.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis. Oncol. 2017, 2017, PO.17.00073. [Google Scholar] [CrossRef]
- Loo, S.K.; Ab Hamid, S.S.; Musa, M.; Wong, K.K. DNMT1 is associated with cell cycle and DNA replication gene sets in diffuse large B-cell lymphoma. Pathol. Res. Pract. 2018, 214, 134–143. [Google Scholar] [CrossRef]
- Maguire, A.; Chen, X.; Wisner, L.; Malasi, S.; Ramsower, C.; Kendrick, S.; Barrett, M.T.; Glinsmann-Gibson, B.; McGrath, M.; Rimsza, L.M. Enhanced DNA repair and genomic stability identify a novel HIV-related diffuse large B-cell lymphoma signature. Int. J. Cancer 2019, 145, 3078–3088. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Li, B.; Ma, R.; Yi, H.; Xu, Z.; Peng, Y.; Yu, D.; Wu, H.; Cheng, T.; Lu, Y.; et al. Anti-DLBCL efficacy of DCZ0825 in vitro and in vivo: Involvement of the PI3K-AKT-mTOR/JNK pathway. Acta Biochim. Biophys. Sin. 2021, 53, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Li, M.; Feng, Y.; Sun, F.; Zhang, L.; Xu, Y.; Lu, S.; Zhu, J.; Huang, J.; Wang, J.; et al. MDM2-P53 Signaling Pathway-Mediated Upregulation of CDC20 Promotes Progression of Human Diffuse Large B-Cell Lymphoma. OncoTargets Ther. 2020, 13, 10475–10487. [Google Scholar] [CrossRef] [PubMed]
- Chatila, W.K.; Kim, J.K.; Walch, H.; Marco, M.R.; Chen, C.T.; Wu, F.; Omer, D.M.; Khalil, D.N.; Ganesh, K.; Qu, X.; et al. Genomic and transcriptomic determinants of response to neoadjuvant therapy in rectal cancer. Nat. Med. 2022, 28, 1646–1655. [Google Scholar] [CrossRef]
- Yang, J.; Lin, Y.; Huang, Y.; Jin, J.; Zou, S.; Zhang, X.; Li, H.; Feng, T.; Chen, J.; Zuo, Z.; et al. Genome landscapes of rectal cancer before and after preoperative chemoradiotherapy. Theranostics 2019, 9, 6856–6866. [Google Scholar] [CrossRef]
- Laumont, C.M.; Banville, A.C.; Gilardi, M.; Hollern, D.P.; Nelson, B.H. Tumour-infiltrating B cells: Immunological mechanisms, clinical impact and therapeutic opportunities. Nat. Rev. Cancer 2022, 22, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Fu, G.; Huang, M.; Kao, X.; Zhu, J.; Dai, Z.; Chen, Y.; Li, H.; Zhou, J.; Chu, X.; et al. scRNA-seq of colorectal cancer shows regional immune atlas with the function of CD20(+) B cells. Cancer Lett. 2024, 584, 216664. [Google Scholar] [CrossRef]
- Xu, Y.; Wei, Z.; Feng, M.; Zhu, D.; Mei, S.; Wu, Z.; Feng, Q.; Chang, W.; Ji, M.; Liu, C.; et al. Tumor-infiltrated activated B cells suppress liver metastasis of colorectal cancers. Cell Rep. 2022, 40, 111295. [Google Scholar] [CrossRef]
- Martin, A.L.; Powell, C.; Nagy, M.Z.; Innamarato, P.; Powers, J.; Nichols, D.; Anadon, C.M.; Chaurio, R.A.; Kim, S.; Wang, M.H.; et al. Anti-4-1BB immunotherapy enhances systemic immune effects of radiotherapy to induce B and T cell-dependent anti-tumor immune activation and improve tumor control at unirradiated sites. Cancer Immunol. Immunother. 2023, 72, 1445–1460. [Google Scholar] [CrossRef]
- Cho, H.; Kim, J.E.; Hong, Y.S.; Kim, S.Y.; Kim, J.; Ryu, Y.M.; Kim, S.Y.; Kim, T.W. Comprehensive evaluation of the tumor immune microenvironment and its dynamic changes in patients with locally advanced rectal cancer treated with preoperative chemoradiotherapy: From the phase II ADORE study. Oncoimmunology 2022, 11, 2148374. [Google Scholar] [CrossRef]
- Goff, P.H.; Riolobos, L.; LaFleur, B.J.; Spraker, M.B.; Seo, Y.D.; Smythe, K.S.; Campbell, J.S.; Pierce, R.H.; Zhang, Y.; He, Q.; et al. Neoadjuvant Therapy Induces a Potent Immune Response to Sarcoma, Dominated by Myeloid and B Cells. Clin. Cancer Res. 2022, 28, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Feng, X.; Chen, B.; Wu, Y.; Wang, X.; Tang, Z.; Huang, M.; Guo, X. CDCA genes as prognostic and therapeutic targets in Colon adenocarcinoma. Hereditas 2025, 162, 19. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Zhang, Z.; Peng, W.; Li, J.; Gu, Q.; Ji, D.; Wang, Q.; Zhang, Y.; Ji, B.; Wang, S.; et al. CDCA3 mediates p21-dependent proliferation by regulating E2F1 expression in colorectal cancer. Int. J. Oncol. 2018, 53, 2021–2033. [Google Scholar] [CrossRef]
- Shen, A.; Liu, L.; Chen, H.; Qi, F.; Huang, Y.; Lin, J.; Sferra, T.J.; Sankararaman, S.; Wei, L.; Chu, J.; et al. Cell division cycle associated 5 promotes colorectal cancer progression by activating the ERK signaling pathway. Oncogenesis 2019, 8, 19. [Google Scholar] [CrossRef]
- Gao, J.; Wang, H.; Qiu, X.; Tang, J. E2F3/CDCA2 reduces radiosensitivity in gastric adenocarcinoma by activating PI3K/AKT pathway. Br. J. Radiol. 2023, 96, 20230477. [Google Scholar] [CrossRef]
- Feng, Y.; Qian, W.; Zhang, Y.; Peng, W.; Li, J.; Gu, Q.; Ji, D.; Zhang, Z.; Wang, Q.; Zhang, D.; et al. CDCA2 promotes the proliferation of colorectal cancer cells by activating the AKT/CCND1 pathway in vitro and in vivo. BMC Cancer 2019, 19, 576. [Google Scholar] [CrossRef]
- Fang, H.; Sheng, S.; Chen, B.; Wang, J.; Mao, D.; Han, Y.; Liu, Y.; Wang, X.; Gui, S.; Zhang, T.; et al. A Pan-Cancer Analysis of the Oncogenic Role of Cell Division Cycle-Associated Protein 4 (CDCA4) in Human Tumors. Front. Immunol. 2022, 13, 826337. [Google Scholar] [CrossRef]
- Xu, C.; Cao, H.; Sui, Y.; Zhang, H.; Shi, C.; Wu, J.; Ma, R.; Feng, J. CDCA4 suppresses epithelial-mesenchymal transtion (EMT) and metastasis in Non-small cell lung cancer through modulating autophagy. Cancer Cell Int. 2021, 21, 48. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wu, X.; Li, F.; Huang, D.; Zhu, W. CDCA4, a downstream gene of the Nrf2 signaling pathway, regulates cell proliferation and apoptosis in the MCF-7/ADM human breast cancer cell line. Mol. Med. Rep. 2018, 17, 1507–1512. [Google Scholar] [CrossRef]
- Li, J.; Zhang, F.; Li, H.; Peng, F.; Wang, Z.; Peng, H.; He, J.; Li, Y.; He, L.; Wei, L. Circ_0010220-mediated miR-503-5p/CDCA4 axis contributes to osteosarcoma progression tumorigenesis. Gene 2020, 763, 145068. [Google Scholar] [CrossRef]
- Gentilini, A.; Pastore, M.; Marra, F.; Raggi, C. The Role of Stroma in Cholangiocarcinoma: The Intriguing Interplay between Fibroblastic Component, Immune Cell Subsets and Tumor Epithelium. Int. J. Mol. Sci. 2018, 19, 2885. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef]
- Ke, Z.; Wang, C.; Wu, T.; Wang, W.; Yang, Y.; Dai, Y. PAR2 deficiency enhances myeloid cell-mediated immunosuppression and promotes colitis-associated tumorigenesis. Cancer Lett. 2020, 469, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wei, T.; Ma, K.; Zhang, J.; Lu, J.; Zhao, J.; Huang, J.; Zeng, T.; Xie, Y.; Liang, Y.; et al. Single-cell RNA sequencing highlights epithelial and microenvironmental heterogeneity in malignant progression of pancreatic ductal adenocarcinoma. Cancer Lett. 2024, 584, 216607. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Li, Y.; Yang, L.; Zhang, Z.; Guo, D.; Zhang, J.; Lu, Y. Profiling transcriptional heterogeneity of epithelium, fibroblasts, and immune cells in esophageal squamous cell carcinoma by single-cell RNA sequencing. FASEB J. 2022, 36, e22620. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Gao, Y.; Hu, Z.; Si, J.; Zhang, Y.; Cai, Q. Identifying CDCA4 as a Radiotherapy Resistance-Associated Gene in Colorectal Cancer by an Integrated Bioinformatics Analysis Approach. Genes 2025, 16, 696. https://doi.org/10.3390/genes16060696
Chen L, Gao Y, Hu Z, Si J, Zhang Y, Cai Q. Identifying CDCA4 as a Radiotherapy Resistance-Associated Gene in Colorectal Cancer by an Integrated Bioinformatics Analysis Approach. Genes. 2025; 16(6):696. https://doi.org/10.3390/genes16060696
Chicago/Turabian StyleChen, Lin, Yawei Gao, Zhiqing Hu, Jingwen Si, Yuchao Zhang, and Qingping Cai. 2025. "Identifying CDCA4 as a Radiotherapy Resistance-Associated Gene in Colorectal Cancer by an Integrated Bioinformatics Analysis Approach" Genes 16, no. 6: 696. https://doi.org/10.3390/genes16060696
APA StyleChen, L., Gao, Y., Hu, Z., Si, J., Zhang, Y., & Cai, Q. (2025). Identifying CDCA4 as a Radiotherapy Resistance-Associated Gene in Colorectal Cancer by an Integrated Bioinformatics Analysis Approach. Genes, 16(6), 696. https://doi.org/10.3390/genes16060696