
Genetic Variation and Metapopulation Structure Inform Recovery Goals in a Threatened Species

Abstract
1. Introduction
2. Materials and Methods
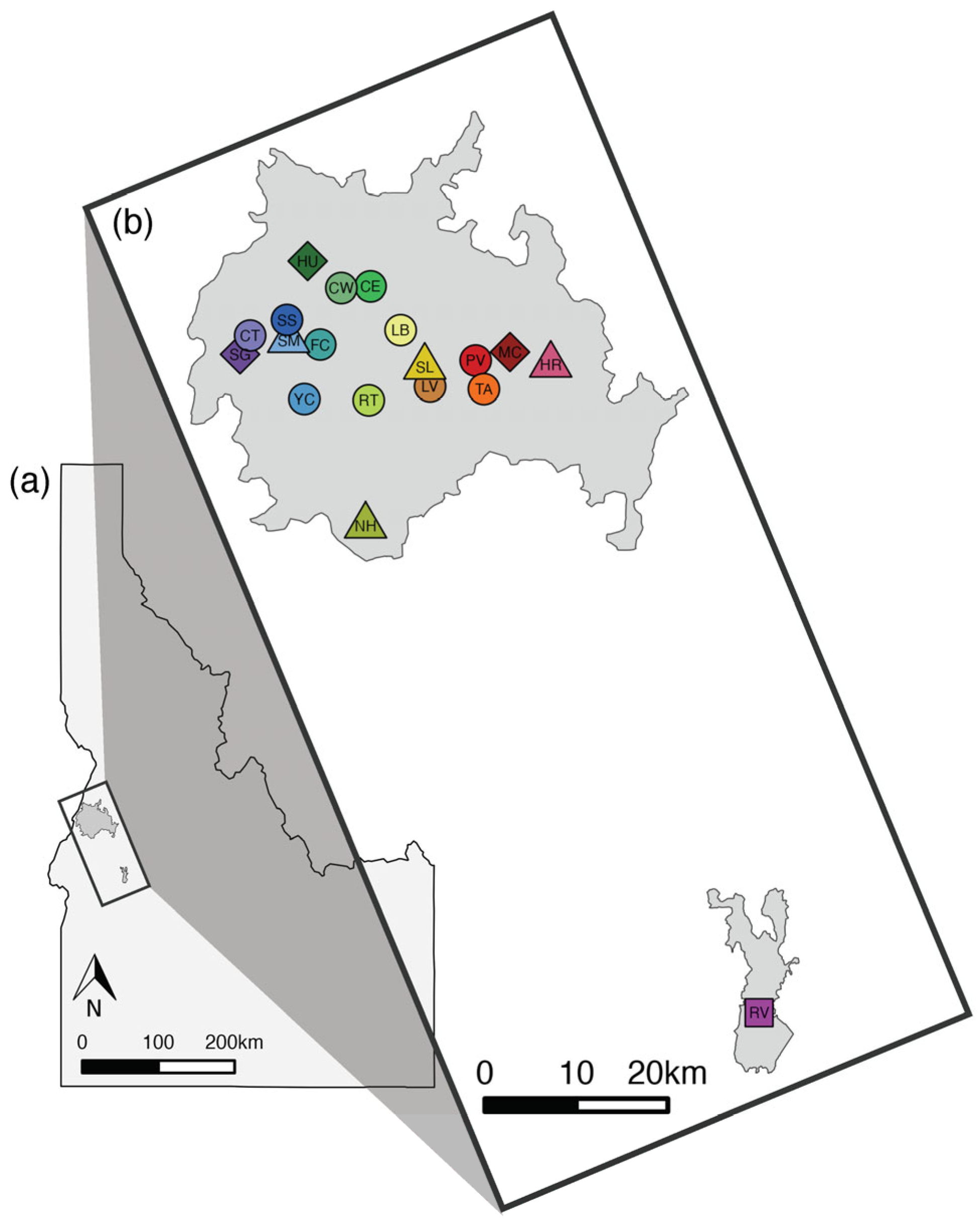
2.1. Study Area and Sample Collection
2.2. GT-seq Library Preparation, Sequencing, and Genotyping
2.3. Evolutionarily Significant Units
2.4. Management Units
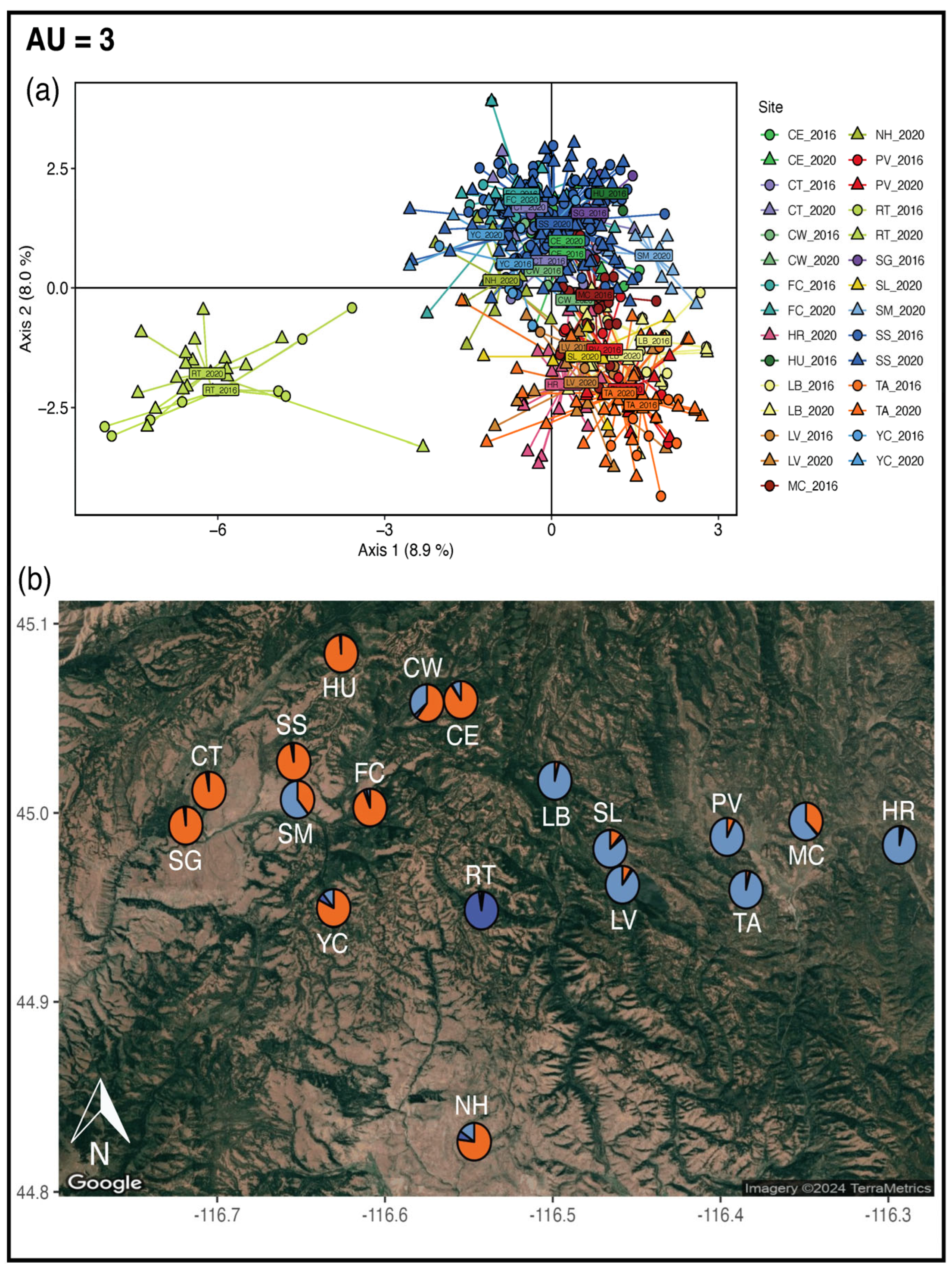
2.5. Adaptive Units
2.6. The Spatially Disjunct Population of Round Valley
3. Results
3.1. Genotyping
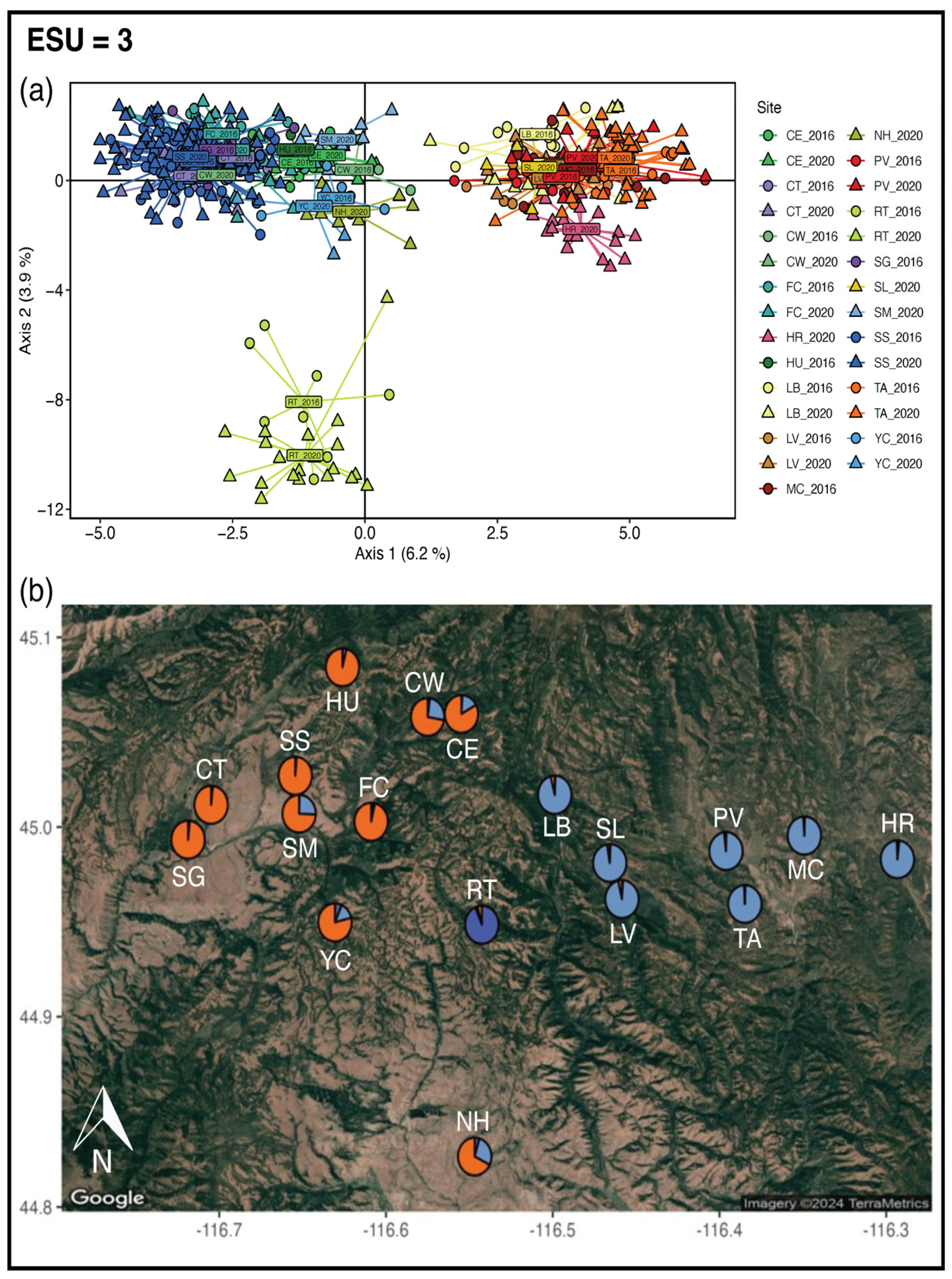
3.2. Evolutionarily Significant Units
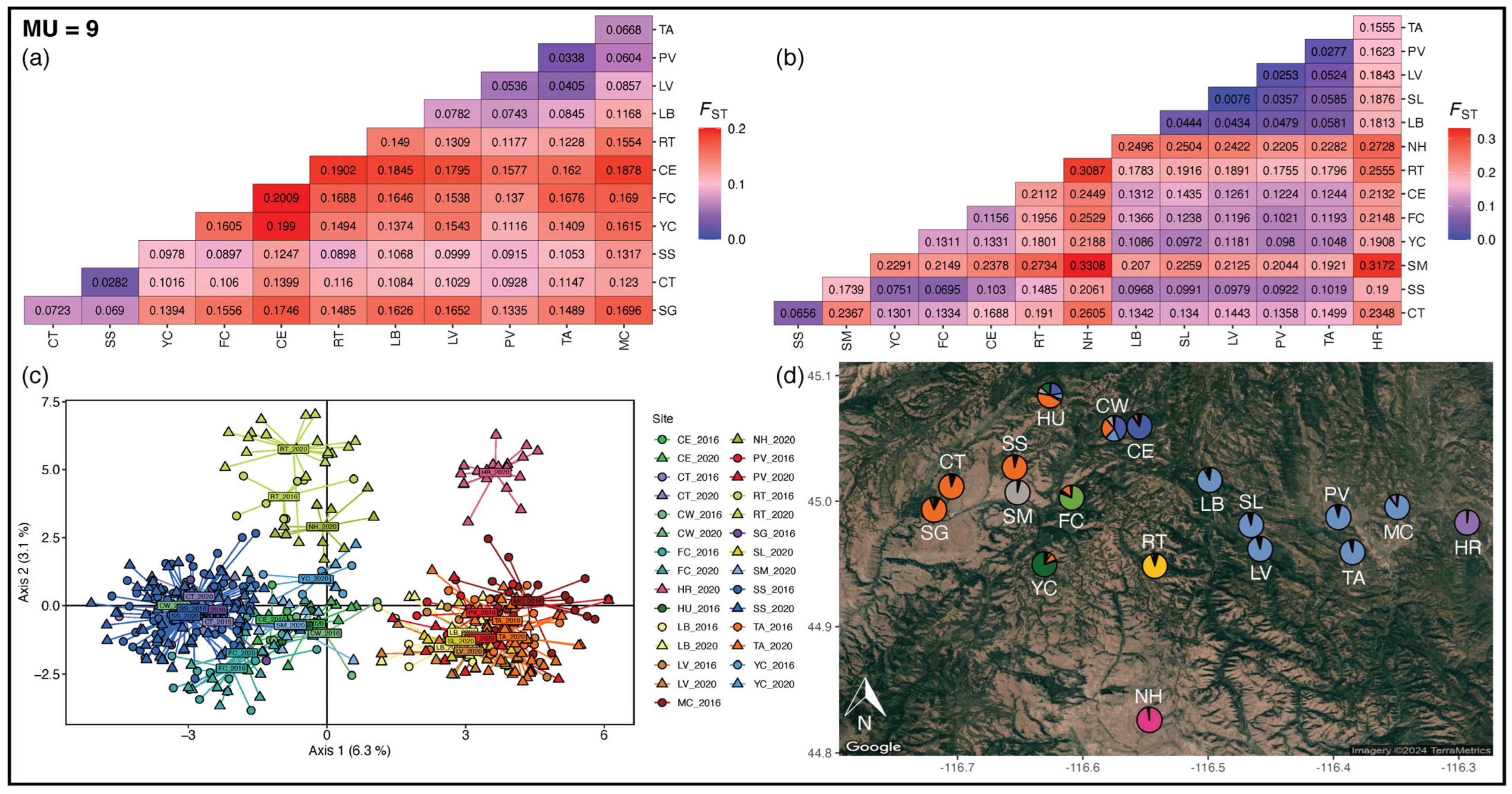
3.3. Management Units
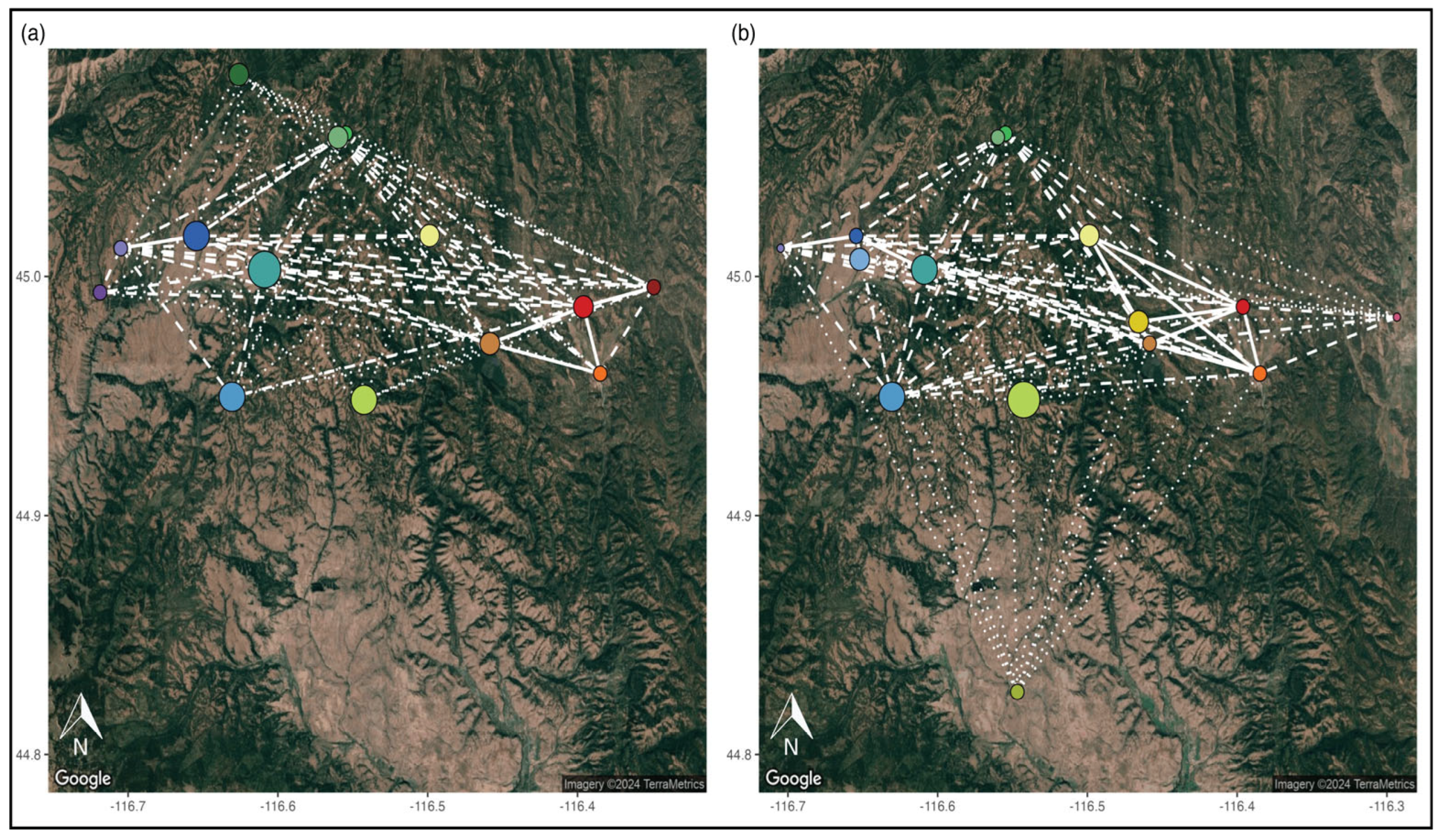
3.4. Adaptive Units
3.5. The Distant Population of Round Valley
4. Discussion
4.1. Evolutionarily Significant Units
4.2. Management Units
4.3. Adaptive Units
4.4. Management and Conservation Implications
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allendorf, F.W. Genetics and the conservation of natural populations: Allozymes to genomes. Mol. Ecol. 2017, 26, 420–430. [Google Scholar] [CrossRef]
- Allendorf, F.W.; Funk, W.C.; Aitken, S.N.; Byrne, M.; Luikart, G. Conservation and the Genomics of Populations; Oxford University Press: Oxford, UK, 2022; ISBN 978-0-19-259857-8. [Google Scholar]
- Allendorf, F.W.; Luikart, G.H.; Aitken, S.N. Conservation and the Genetics of Populations; Wiley-Blackwell: Malden, MA, USA, 2012. [Google Scholar]
- Frankham, R. Inbreeding in the wild really does matter. Heredity 2010, 104, 124. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, E.; Sirén, J.; Somervuo, P.; Ruokolainen, L.; Ovaskainen, O.; Hanski, I. Scaling up the effects of inbreeding depression from individuals to metapopulations. J. Anim. Ecol. 2019, 88, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Palstra, F.P.; Ruzzante, D.E. Genetic estimates of contemporary effective population size: What can they tell us about the importance of genetic stochasticity for wild population persistence? Mol. Ecol. 2008, 17, 3428–3447. [Google Scholar] [CrossRef]
- Waples, R.S. What is Ne, anyway? J. Hered. 2022, 113, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, P.W.; Gilpin, M.E. Genetic effective size of a metapopulation. In Metapopulation Biology; Hanski, I., Gilpin, M.E., Eds.; Academic Press: San Diego, CA, USA, 1997; pp. 165–181. ISBN 978-0-12-323445-2. [Google Scholar]
- Kurland, S.; Ryman, N.; Hössjer, O.; Laikre, L. Effects of subpopulation extinction on effective size (Ne) of metapopulations. Conserv. Genet. 2023, 24, 417–433. [Google Scholar] [CrossRef]
- Ryman, N.; Laikre, L.; Hössjer, O. Variance effective population size is affected by census size in sub-structured populations. Mol. Ecol. Resour. 2023, 23, 1334–1347. [Google Scholar] [CrossRef]
- Frankham, R. Genetics and extinction. Biol. Conserv. 2005, 126, 131–140. [Google Scholar] [CrossRef]
- Lande, R. Risks of population extinction from demographic and environmental stochasticity and random catastrophes. Am. Nat. 1993, 142, 911–927. [Google Scholar] [CrossRef]
- Baalsrud, H.T.; Sæther, B.E.; Hagen, I.J.; Myhre, A.M.; Ringsby, T.H.; Pärn, H.; Jensen, H. Effects of population characteristics and structure on estimates of effective population size in a house sparrow metapopulation. Mol. Ecol. 2014, 23, 2653–2668. [Google Scholar] [CrossRef]
- Hastings, A.; Harrison, S. Metapopulation dynamics and genetics. Annu. Rev. Ecol. Syst. 1994, 25, 167–188. [Google Scholar] [CrossRef]
- Hohenlohe, P.A.; Funk, W.C.; Rajora, O.P. Population genomics for wildlife conservation and management. Mol. Ecol. 2021, 30, 62–82. [Google Scholar] [CrossRef]
- Walters, A.D.; Schwartz, M.K. Population genomics for the management of wild vertebrate populations. In Population Genomics: Wildlife; Hohenlohe, P.A., Rajora, O.P., Eds.; Springer: Cham, Switzerland, 2020; pp. 419–436. [Google Scholar]
- Campbell, N.R.; Harmon, S.A.; Narum, S.R. Genotyping-in-thousands by sequencing (GT-Seq): A cost effective SNP genotyping method based on custom amplicon sequencing. Mol. Ecol. Resour. 2015, 15, 855–867. [Google Scholar] [CrossRef]
- Burgess, B.T.; Irvine, R.L.; Russello, M.A. A Genotyping-in-thousands by sequencing panel to inform invasive deer management using noninvasive fecal and hair samples. Ecol. Evol. 2022, 12, e8993. [Google Scholar] [CrossRef]
- Hayward, K.M.; Clemente-Carvalho, R.B.G.; Jensen, E.L.; de Groot, P.V.C.; Branigan, M.; Dyck, M.; Tschritter, C.; Sun, Z.; Lougheed, S.C. Genotyping-in-thousands by sequencing (GT-Seq) of noninvasive faecal and degraded samples: A new panel to enable ongoing monitoring of Canadian polar bear populations. Mol. Ecol. Resour. 2022, 22, 1906–1918. [Google Scholar] [CrossRef]
- Schmidt, D.A.; Campbell, N.R.; Govindarajulu, P.; Larsen, K.W.; Russello, M.A. Genotyping-in-thousands by sequencing (GT-Seq) panel development and application to minimally invasive DNA samples to support studies in molecular ecology. Mol. Ecol. Resour. 2020, 20, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, S.; Mestre, F.; White, T.A.; Paupério, J.; Alves, P.C.; Searle, J.B. Integrative approaches to guide conservation decisions: Using genomics to define conservation units and functional corridors. Mol. Ecol. 2018, 27, 3452–3465. [Google Scholar] [CrossRef] [PubMed]
- Boussarie, G.; Momigliano, P.; Robbins, W.D.; Bonnin, L.; Cornu, J.-F.; Fauvelot, C.; Kiszka, J.J.; Manel, S.; Mouillot, D.; Vigliola, L. Identifying barriers to gene flow and hierarchical conservation units from seascape genomics: A modelling framework applied to a marine predator. Ecography 2022, 2022, e06158. [Google Scholar] [CrossRef]
- Forester, B.R.; Murphy, M.; Mellison, C.; Petersen, J.; Pilliod, D.S.; Van Horne, R.; Harvey, J.; Funk, W.C. Genomics-informed delineation of conservation units in a desert amphibian. Mol. Ecol. 2022, 31, 5249–5269. [Google Scholar] [CrossRef]
- Weckworth, B.V.; Hebblewhite, M.; Mariani, S.; Musiani, M. Lines on a map: Conservation units, meta-population dynamics, and recovery of woodland caribou in Canada. Ecosphere 2018, 9, e02323. [Google Scholar] [CrossRef]
- Waples, R.S. Pacific Salmon, Oncorhynchus Spp., and the definition of “species” under the Endangered Species Act. Mar. Fish. Rev. 1991, 53, 11. [Google Scholar]
- Funk, W.C.; McKay, J.K.; Hohenlohe, P.A.; Allendorf, F.W. Harnessing genomics for delineating conservation units. Trends Ecol. Evol. 2012, 27, 489–496. [Google Scholar] [CrossRef]
- Moritz, C. Defining ‘Evolutionarily Significant Units’ for conservation. Trends Ecol. Evol. 1994, 9, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Palsboll, P.; Berube, M.; Allendorf, F. Identification of management units using population genetic data. Trends Ecol. Evol. 2007, 22, 11–16. [Google Scholar] [CrossRef]
- Turbek, S.P.; Funk, W.C.; Ruegg, K.C. Where to draw the line? Expanding the delineation of conservation units to highly mobile taxa. J. Hered. 2023, 114, 300–311. [Google Scholar] [CrossRef] [PubMed]
- De Guia, A.P.O.; Saitoh, T. The gap between the concept and definitions in the Evolutionarily Significant Unit: The need to integrate neutral genetic variation and adaptive variation. Ecol. Res. 2007, 22, 604–612. [Google Scholar] [CrossRef]
- Moritz, C. Conservation units and translocations: Strategies for conserving evolutionary processes. Hereditas 1999, 130, 217–228. [Google Scholar] [CrossRef]
- USFWS. Recovery Plan for the Northern Idaho Ground Squirrel (Spermophilus Brunneus Brunneus); U.S. Fish and Wildlife Service: Portland, OR, USA, 2003. [Google Scholar]
- Wagner, B.; Evans Mack, D. Long-Term Population Monitoring of Northern Idaho Ground Squirrel: 2020 Implementation and Population Estimates; Idaho Department of Fish and Game: Boise, ID, USA, 2020. [Google Scholar]
- Allison, A.Z.; Conway, C.J.; Morris, A.E. Why hibernate? Tests of four hypotheses to explain intraspecific variation in hibernation phenology. Funct. Ecol. 2023, 37, 1580–1593. [Google Scholar] [CrossRef]
- Goldberg, A.R.; Conway, C.J. Hibernation behavior of a federally threatened ground squirrel: Climate change and habitat selection implications. J. Mammal. 2021, 102, 574–587. [Google Scholar] [CrossRef]
- Gavin, T.A.; Sherman, P.W.; Yensen, E.; May, B. Population genetic structure of the northern Idaho ground squirrel (Spermophilus brunneus brunneus). J. Mammal. 1999, 80, 156–168. [Google Scholar] [CrossRef]
- Helmstetter, N.A.; Conway, C.J.; Stevens, B.S.; Goldberg, A.R. Balancing transferability and complexity of species distribution models for rare species conservation. Divers. Distrib. 2021, 27, 95–108. [Google Scholar] [CrossRef]
- Sherman, P.W.; Runge, M.C. Demography of a population collapse: The northern Idaho ground squirrel (Spermophilus brunneus brunneus). Ecology 2002, 83, 2816–2831. [Google Scholar] [CrossRef]
- Suronen, E.F.; Newingham, B.A. Restoring habitat for the northern Idaho ground squirrel (Urocitellus brunneus brunneus): Effects of prescribed burning on dwindling habitat. For. Ecol. Manag. 2013, 304, 224–232. [Google Scholar] [CrossRef]
- Yensen, E.; Dyni, E.J. Why is the northern Idaho ground squirrel rare? Northwest Sci. 2020, 94, 1–23. [Google Scholar] [CrossRef]
- Dyni, E.J.; Yensen, E. Dietary similarity in sympatric Idaho and Columbian ground squirrels (Spermophilus brunneus and S. columbianus). Northwest Sci. 1996, 70, 99–108. [Google Scholar]
- Barbosa, S.; Andrews, K.R.; Goldberg, A.R.; Gour, D.S.; Hohenlohe, P.A.; Conway, C.J.; Waits, L.P. The role of neutral and adaptive genomic variation in population diversification and speciation in two ground squirrel species of conservation concern. Mol. Ecol. 2021, 30, 4673–4694. [Google Scholar] [CrossRef]
- Garner, A.; Rachlow, J.L.; Waits, L.P. Genetic diversity and population divergence in fragmented habitats: Conservation of Idaho ground squirrels. Conserv. Genet. 2005, 6, 759–774. [Google Scholar] [CrossRef]
- Goldberg, A.R.; Conway, C.J.; Biggins, D.E. Flea sharing among sympatric rodent hosts: Implications for potential plague effects on a threatened sciurid. Ecosphere 2020, 11, e03033. [Google Scholar] [CrossRef]
- Goldberg, A.R.; Conway, C.J.; Biggins, D.E. Effects of experimental flea removal and plague vaccine treatments on survival of northern Idaho ground squirrels and two coexisting sciurids. Glob. Ecol. Conserv. 2021, 26, e01489. [Google Scholar] [CrossRef]
- Gill, A.E.; Yensen, E. Biochemical differentiation in the Idaho ground squirrel, Spermophilus brunneus (Rodentia: Sciuridae). Great Basin Nat. 1992, 52, 155–159. [Google Scholar]
- Hoisington-Lopez, J.L.; Waits, L.P.; Sullivan, J. Species limits and integrated taxonomy of the Idaho ground squirrel (Urocitellus brunneus): Genetic and ecological differentiation. J. Mammal. 2012, 93, 589–604. [Google Scholar] [CrossRef]
- Garrett, M.J.; Nerkowski, S.A.; Kieran, S.; Campbell, N.R.; Barbosa, S.; Conway, C.J.; Hohenlohe, P.A.; Waits, L.P. Development and validation of a GT-Seq panel for genetic monitoring in a threatened species using minimally invasive sampling. Ecol. Evol. 2024, 14, e11321. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- Goudet, J. Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.; O’Hara, R.; Simpson, G.; Solymos, P.; et al. vegan: Community Ecology Package, Version 2.4-3; CRAN: WU Wien, Austria, 2017; Available online: https://vegandevs.github.io/vegan/ (accessed on 4 June 2024).
- Chessel, D.; Dufour, A.B.; Thioulouse, J. The Ade4 Package—I: One-Table Methods. R News 2004, 4, 5–10. [Google Scholar]
- Kassambara, A. ggpubr: “ggplot2” Based Publication Ready Plots; R Package Version 0.6.0 2023; CRAN: WU Wien, Austria, 2023; Available online: https://rpkgs.datanovia.com/ggpubr/ (accessed on 4 June 2024).
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Yu, G. scatterpie: Scatter Pie Plot; R Package Version 0.2.1; CRAN: WU Wien, Austria, 2021; Available online: https://cran.r-project.org/package=scatterpie (accessed on 4 June 2024).
- Dyer, R.J.; Nason, J.D. Population graphs: The graph theoretic shape of genetic structure. Mol. Ecol. 2004, 13, 1713–1727. [Google Scholar] [CrossRef]
- Balkenhol, N.; Cushman, S.; Storfer, A.; Waits, L. Landscape Genetics: Concepts, Methods, Applications; John Wiley & Sons: Hoboken, NJ, USA, 2015; ISBN 978-1-118-52529-6. [Google Scholar]
- Billerman, S.M.; Jesmer, B.R.; Watts, A.G.; Schlichting, P.E.; Fortin, M.J.; Funk, W.C.; Hapeman, P.; Muths, E.; Murphy, M.A. Testing theoretical metapopulation conditions with genotypic data from boreal chorus frogs (Pseudacris maculata). Can. J. Zool. 2019, 97, 1042–1053. [Google Scholar] [CrossRef]
- Dale, M.R.T.; Fortin, M.J. From graphs to spatial graphs. Annu. Rev. Ecol. Evol. Syst. 2010, 41, 21–38. [Google Scholar] [CrossRef]
- Garroway, C.J.; Bowman, J.; Carr, D.; Wilson, P.J. Applications of graph theory to landscape genetics. Evol. Appl. 2008, 1, 620–630. [Google Scholar] [CrossRef]
- Miles, L.S.; Dyer, R.J.; Verrelli, B.C. Urban hubs of connectivity: Contrasting patterns of gene flow within and among cities in the western black widow spider. Proc. R. Soc. B Biol. Sci. 2018, 285, 20181224. [Google Scholar] [CrossRef]
- Do, C.; Waples, R.S.; Peel, D.; Macbeth, G.M.; Tillett, B.J.; Ovenden, J.R. NeEstimator v2: Re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 2014, 14, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Wright, S. Evolution and the Genetics of Populations, Volume 4: Variability Within and Among Natural Populations; University of Chicago Press: Chicago, IL, USA, 1984; ISBN 978-0-226-91041-3. [Google Scholar]
- Zero, V.H.; Barocas, A.; Jochimsen, D.M.; Pelletier, A.; Giroux-Bougard, X.; Trumbo, D.R.; Castillo, J.A.; Mack, D.E.; Linnell, M.A.; Pigg, R.M.; et al. Complementary network-based approaches for exploring genetic structure and functional connectivity in two vulnerable, endemic ground squirrels. Front. Genet. 2017, 8, 81. [Google Scholar] [CrossRef]
- Hoisington-Lopez, J. Conservation Genetics, Landscape Genetics and Systematics of the Two Subspecies of the Endemic Idaho Ground Squirrel (Spermophilus brunneus). Master’s Thesis, University of Idaho, Moscow, ID, USA, 2007. [Google Scholar]
- Lowe, W.H.; Allendorf, F.W. What can genetics tell us about population connectivity? Mol. Ecol. 2010, 19, 3038–3051. [Google Scholar] [CrossRef]
- Perez, M.F.; Franco, F.F.; Bombonato, J.R.; Bonatelli, I.A.S.; Khan, G.; Romeiro-Brito, M.; Fegies, A.C.; Ribeiro, P.M.; Silva, G.A.R.; Moraes, E.M. Assessing population structure in the face of isolation by distance: Are we neglecting the problem? Divers. Distrib. 2018, 24, 1883–1889. [Google Scholar] [CrossRef]
- Hanski, I.; Pakkala, T.; Kuussaari, M.; Lei, G. Metapopulation persistence of an endangered butterfly in a fragmented landscape. Oikos 1995, 72, 21–28. [Google Scholar] [CrossRef]
- Clarke, S.H.; Lawrence, E.R.; Matte, J.-M.; Gallagher, B.K.; Salisbury, S.J.; Michaelides, S.N.; Koumrouyan, R.; Ruzzante, D.E.; Grant, J.W.A.; Fraser, D.J. Global assessment of effective population sizes: Consistent taxonomic differences in meeting the 50/500 rule. Mol. Ecol. 2024, 33, e17353. [Google Scholar] [CrossRef]
- Frankham, R.; Bradshaw, C.J.A.; Brook, B.W. Genetics in conservation management: Revised recommendations for the 50/500 rules, red list criteria and population viability analyses. Biol. Conserv. 2014, 170, 56–63. [Google Scholar] [CrossRef]
- Forester, B.R.; Beever, E.A.; Darst, C.; Szymanski, J.; Funk, W.C. Linking evolutionary potential to extinction risk: Applications and future directions. Front. Ecol. Env. 2022, 20, 507–515. [Google Scholar] [CrossRef]
- Yensen, E.; Sherman, P.W. Spermophilus brunneus. Mamm. Species 1997, 560, 1–5. [Google Scholar] [CrossRef]
- Allison, A.Z.T.; Conway, C.J.; Goldberg, A.R. Weather influences survival probability in two coexisting mammals directly and indirectly via competitive asymmetry. Ecology 2024, 105, e4229. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.; Hakanson, E.C.; Conway, C.J. Effects of Forest Encroachment on Behavior and Demography of the Northern Idaho Ground Squirrel: Annual Progress Report 2023; Wildlife Research Report #2023-02; Idaho Cooperative Fish and Wildlife Research Unit: Moscow, ID, USA, 2023. [Google Scholar]
- Abatzoglou, J.T.; Marshall, A.M.; Harley, G.L. Observed and Projected Changes in Idaho’s Climate; Idaho Climate-Economy Impacts Assessment; James A. & Louise McClure Center for Public Policy Research, University of Idaho: Boise, ID, USA, 2021. [Google Scholar]
- Fitzpatrick, S.W.; Gerberich, J.C.; Angeloni, L.M.; Bailey, L.L.; Broder, E.D.; Torres-Dowdall, J.; Handelsman, C.A.; López-Sepulcre, A.; Reznick, D.N.; Ghalambor, C.K.; et al. Gene flow from an adaptively divergent source causes rescue through genetic and demographic factors in two wild populations of Trinidadian guppies. Evol. Appl. 2016, 9, 879–891. [Google Scholar] [CrossRef]
- Seaborn, T.; Andrews, K.R.; Applestein, C.V.; Breech, T.M.; Garrett, M.J.; Zaiats, A.; Caughlin, T.T. Integrating genomics in population models to forecast translocation success. Restor. Ecol. 2021, 29, e13395. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site Name | Site Abbr. | Year | N | Observed Heterozygosity | Expected Heterozygosity | Private Alleles | Degree | Betweeness | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All SNPs | Neutral SNPs | Adaptive SNPs | All SNPs | Neutral SNPs | Adaptive SNPs | |||||||
| Cold Springs East | CE | 2016 | 10 | 0.270 | 0.308 | 0.174 | 0.254 | 0.285 | 0.173 | 0 | 4 | 14 |
| 2020 | 12 | 0.292 | 0.323 | 0.210 | 0.275 | 0.308 | 0.192 | 0 | 4 | 12 | ||
| Cap Gun/Tree Valley | CT | 2016 | 6 | 0.291 | 0.325 | 0.201 | 0.303 | 0.345 | 0.194 | 0 | 4 | 5 |
| 2020 | 8 | 0.274 | 0.308 | 0.186 | 0.278 | 0.314 | 0.187 | 0 | 4 | 9 | ||
| Cold Springs West | CW | 2016 | 4 | 0.291 | 0.311 | 0.238 | 0.302 | 0.319 | 0.261 | 0 | 6 | 14 |
| 2020 | 1 | 0.305 | 0.358 | 0.171 | NA | NA | NA | 0 | 2 | 0 | ||
| Fawn Creek | FC | 2016 | 15 | 0.265 | 0.287 | 0.208 | 0.258 | 0.279 | 0.203 | 0 | 10 | 22 |
| 2020 | 19 | 0.297 | 0.317 | 0.245 | 0.283 | 0.302 | 0.233 | 0 | 8 | 29 | ||
| Hot Springs Road | HR | 2020 | 19 | 0.294 | 0.315 | 0.240 | 0.280 | 0.299 | 0.229 | 0 | 2 | 0 |
| Huckleberry | HU | 2016 | 2 | 0.309 | 0.363 | 0.174 | 0.208 | 0.242 | 0.119 | 0 | 6 | 0 |
| Lower Butter | LB | 2016 | 12 | 0.334 | 0.370 | 0.240 | 0.291 | 0.320 | 0.213 | 0 | 6 | 14 |
| 2020 | 15 | 0.327 | 0.351 | 0.266 | 0.314 | 0.334 | 0.262 | 0 | 6 | 14 | ||
| Lost Valley | LV | 2016 | 10 | 0.298 | 0.332 | 0.211 | 0.302 | 0.337 | 0.213 | 0 | 6 | 30 |
| 2020 | 16 | 0.308 | 0.333 | 0.244 | 0.311 | 0.339 | 0.238 | 0 | 4 | 33 | ||
| Mud Creek | MC | 2016 | 22 | 0.308 | 0.356 | 0.179 | 0.301 | 0.345 | 0.185 | 0 | 4 | 0 |
| North Hornet | NH | 2020 | 10 | 0.229 | 0.243 | 0.192 | 0.240 | 0.264 | 0.177 | 0 | 4 | 0 |
| Price Valley | PV | 2016 | 10 | 0.318 | 0.350 | 0.236 | 0.312 | 0.346 | 0.222 | 0 | 6 | 22 |
| 2020 | 14 | 0.326 | 0.358 | 0.245 | 0.315 | 0.350 | 0.224 | 0 | 4 | 24 | ||
| Rocky Top | RT | 2016 | 8 | 0.334 | 0.330 | 0.343 | 0.304 | 0.295 | 0.328 | 12 | 8 | 22 |
| 2020 | 20 | 0.314 | 0.298 | 0.353 | 0.278 | 0.269 | 0.303 | 7 | 10 | 34 | ||
| Summit Gulch | SG | 2016 | 7 | 0.266 | 0.320 | 0.125 | 0.275 | 0.323 | 0.151 | 0 | 4 | 1 |
| Slaughter Gulch | SL | 2020 | 7 | 0.308 | 0.343 | 0.217 | 0.303 | 0.331 | 0.230 | 0 | 6 | 40 |
| Smith Mountain | SM | 2020 | 10 | 0.241 | 0.264 | 0.183 | 0.238 | 0.263 | 0.174 | 0 | 6 | 24 |
| Steve’s Creek/Squirrel Valley/Manor | SS | 2016 | 77 | 0.309 | 0.351 | 0.199 | 0.311 | 0.353 | 0.202 | 0 | 8 | 14 |
| 2020 | 74 | 0.302 | 0.343 | 0.193 | 0.308 | 0.351 | 0.195 | 0 | 4 | 13 | ||
| Tamarack | TA | 2016 | 12 | 0.344 | 0.360 | 0.302 | 0.334 | 0.355 | 0.277 | 0 | 4 | 0 |
| 2020 | 39 | 0.336 | 0.362 | 0.270 | 0.333 | 0.358 | 0.265 | 0 | 4 | 13 | ||
| YCC | YC | 2016 | 6 | 0.310 | 0.328 | 0.262 | 0.282 | 0.300 | 0.239 | 1 | 8 | 6 |
| 2020 | 6 | 0.295 | 0.309 | 0.261 | 0.300 | 0.316 | 0.258 | 0 | 8 | 4 | ||
| Management Unit | Year | N | HO—All SNPs | HO—Neutral SNPs | Ne | Lower CI | Upper CI |
|---|---|---|---|---|---|---|---|
| West | 2016 | 92 | 0.304 | 0.347 | 220.9 | 147.9 | 403.7 |
| 2020 | 82 | 0.299 | 0.34 | 120.8 | 69.8 | 305.6 | |
| SM | 2020 | 10 | 0.241 | 0.264 | 18.7 | 8.8 | 92.5 |
| YC | 2016 | 6 | 0.31 | 0.328 | 18.8 | 5 | inf |
| 2020 | 6 | 0.295 | 0.309 | 2.3 | 0.7 | inf | |
| FC | 2016 | 15 | 0.265 | 0.287 | 6.7 | 2.7 | 16.2 |
| 2020 | 19 | 0.297 | 0.317 | 11 | 5.1 | 26.9 | |
| Cold Springs | 2016 | 14 | 0.276 | 0.306 | 14.5 | 9.2 | 26.2 |
| 2020 | 13 | 0.293 | 0.326 | 2.7 | 1.3 | 16.1 | |
| RT | 2016 | 8 | 0.334 | 0.33 | 24.6 | 9.8 | inf |
| 2020 | 20 | 0.314 | 0.298 | 7 | 3.3 | 11.6 | |
| NH | 2020 | 10 | 0.229 | 0.243 | 11.5 | 5.3 | 34.8 |
| East | 2016 | 66 | 0.32 | 0.355 | 50.7 | 36.9 | 74.1 |
| 2020 | 91 | 0.326 | 0.353 | 50.6 | 35.8 | 75.9 | |
| HR | 2020 | 19 | 0.294 | 0.315 | 11.6 | 5.8 | 27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garrett, M.J.; Conway, C.J.; Waits, L.P.; Hohenlohe, P.A. Genetic Variation and Metapopulation Structure Inform Recovery Goals in a Threatened Species. Genes 2025, 16, 694. https://doi.org/10.3390/genes16060694
Garrett MJ, Conway CJ, Waits LP, Hohenlohe PA. Genetic Variation and Metapopulation Structure Inform Recovery Goals in a Threatened Species. Genes. 2025; 16(6):694. https://doi.org/10.3390/genes16060694
Chicago/Turabian StyleGarrett, Molly J., Courtney J. Conway, Lisette P. Waits, and Paul A. Hohenlohe. 2025. "Genetic Variation and Metapopulation Structure Inform Recovery Goals in a Threatened Species" Genes 16, no. 6: 694. https://doi.org/10.3390/genes16060694
APA StyleGarrett, M. J., Conway, C. J., Waits, L. P., & Hohenlohe, P. A. (2025). Genetic Variation and Metapopulation Structure Inform Recovery Goals in a Threatened Species. Genes, 16(6), 694. https://doi.org/10.3390/genes16060694