
A Novel Recurrent 200 kb CRYL1 Deletion Underlies DFNB1A Hearing Loss in Patients from Northwestern Spain

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Novel Deletion Detection, Validation and Characterization
2.3. del(GJB6-D13S1854), del(GJB6-D13S18530) and CRYL1 del(200 kb)insATTATA Deletion Screening
2.4. GJB2 c.35delG Genotyping
3. Results
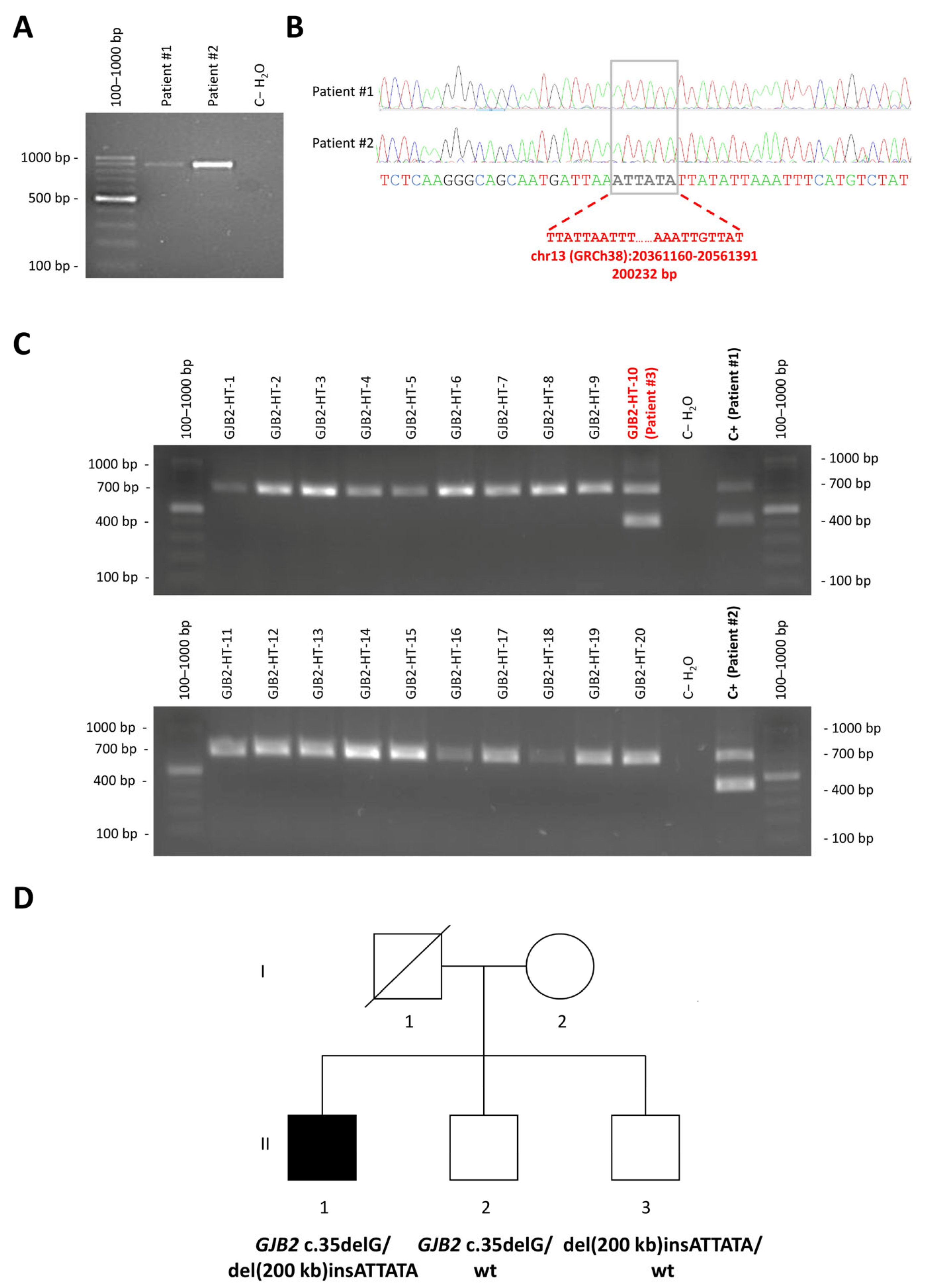
3.1. Detection of a Novel CRYL1 Deletion in Hearing Loss Patients Using an NGS Panel
3.2. Mapping of the Breakpoints of the New CRYL1 Deletion
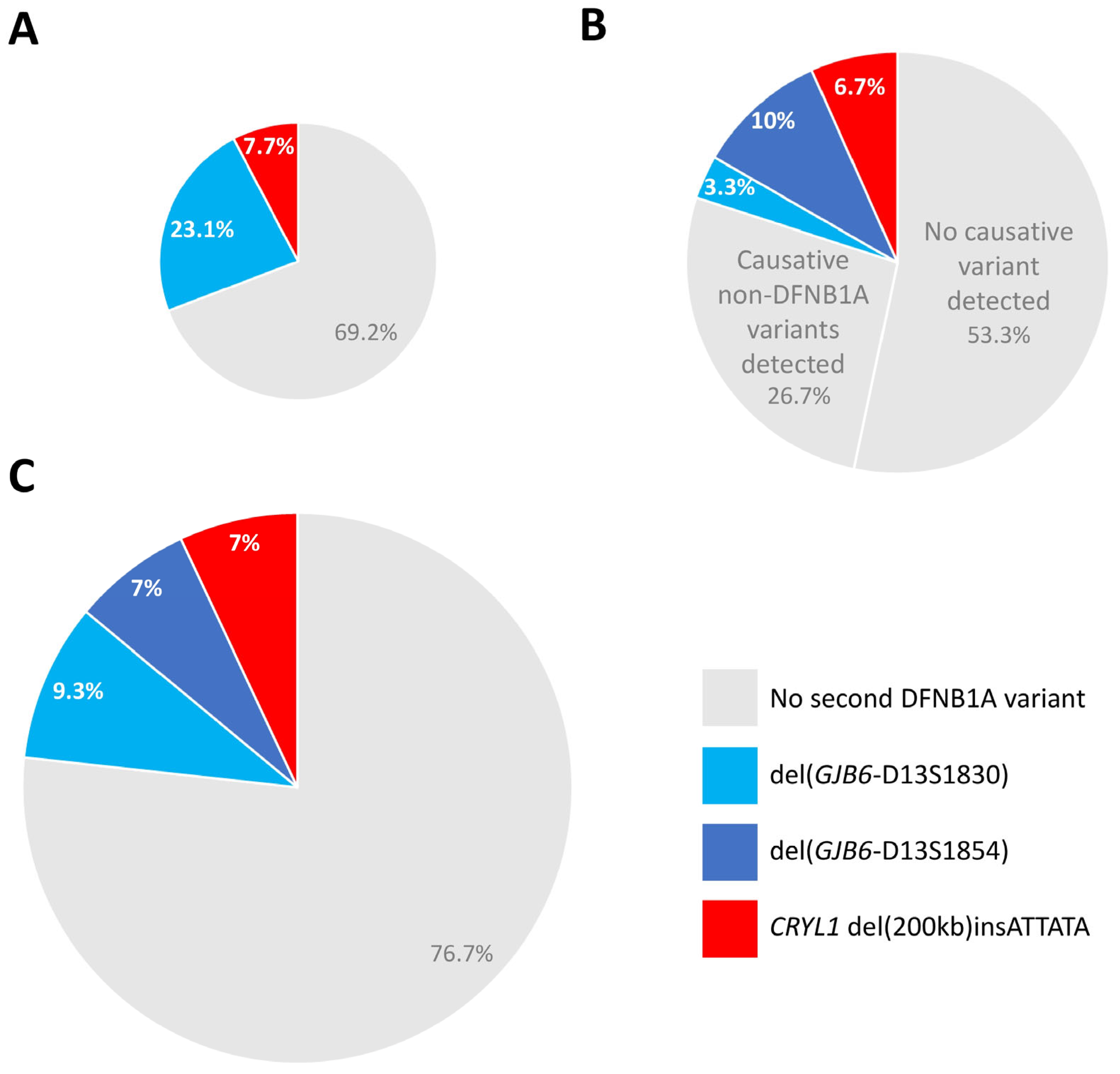
3.3. Novel Deletion Screen in Unexplained Hearing Loss Cases and in the Normal Hearing Population
3.4. Diagnostic Relevance of CRYL1 del(200 kb)insATTATA Within Deaf Patients with Monoallelic Pathogenic GJB2 Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sheffield, A.M.; Smith, R.J.H. The Epidemiology of Deafness. Cold Spring Harb Perspect. Med. 2019, 9, a033258. [Google Scholar] [CrossRef] [PubMed]
- Butcher, E.; Dezateux, C.; Cortina-Borja, M.; Knowles, R.L. Prevalence of permanent childhood hearing loss detected at the universal newborn hearing screen: Systematic review and meta-analysis. PLoS ONE 2019, 14, e0219600. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.B.; Shearer, A.E.; Smith, R.J.H. Genetic sensorineural hearing loss. In Cummings Otolaryngology Head and Neck Surgery; Flint, P.W., Ed.; Elsevier: Philadelphia, PA, USA, 2015; pp. 2285–2300. [Google Scholar]
- Lucotte, G. High prevalences of carriers of the 35delG mutation of connexin 26 in the Mediterranean area. Int. J. Pediatr. Otorhinolaryngol. 2007, 71, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Hoefsloot, L.H.; Roux, A.F.; Bitner-Glindzicz, M. EMQN Best Practice guidelines for diagnostic testing of mutations causing non-syndromic hearing impairment at the DFNB1 locus. Eur. J. Hum. Genet. 2013, 21, 1325–1329. [Google Scholar] [PubMed]
- del Castillo, I.; Villamar, M.; Moreno-Pelayo, M.A.; del Castillo, F.J.; Alvarez, A.; Telleria, D.; Menendez, I.; Moreno, F. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N. Engl. J. Med. 2002, 346, 243–249. [Google Scholar] [CrossRef] [PubMed]
- del Castillo, F.J.; Rodriguez-Ballesteros, M.; Alvarez, A.; Hutchin, T.; Leonardi, E.; de Oliveira, C.A.; Azaiez, H.; Brownstein, Z.; Avenarius, M.R.; Marlin, S.; et al. A novel deletion involving the connexin-30 gene, del(GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J. Med. Genet. 2005, 42, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Wilch, E.; Azaiez, H.; Fisher, R.A.; Elfenbein, J.; Murgia, A.; Birkenhager, R.; Bolz, H.; Da Silva-Costa, S.M.; Del Castillo, I.; Haaf, T.; et al. A novel DFNB1 deletion allele supports the existence of a distant cis-regulatory region that controls GJB2 and GJB6 expression. Clin. Genet. 2010, 78, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Tayoun, A.N.; Mason-Suares, H.; Frisella, A.L.; Bowser, M.; Duffy, E.; Mahanta, L.; Funke, B.; Rehm, H.L.; Amr, S.S. Targeted Droplet-Digital PCR as a Tool for Novel Deletion Discovery at the DFNB1 Locus. Hum. Mutat. 2016, 37, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Xiang, J.; Sun, X.; Song, N.; Liu, X.; Cai, Q.; Yang, J.; Ye, H.; Xu, J.; Zhang, H.; et al. Genome sequencing unveils the role of copy number variants in hearing loss and identifies novel deletions with founder effect in the DFNB1 Locus. Hum. Mutat. 2024, 2024, 9517114. [Google Scholar] [CrossRef] [PubMed]
- Common, J.E.; Bitner-Glindzicz, M.; O’Toole, E.A.; Barnes, M.R.; Jenkins, L.; Forge, A.; Kelsell, D.P. Specific loss of connexin 26 expression in ductal sweat gland epithelium associated with the deletion mutation del(GJB6-D13S1830). Clin. Exp. Dermatol. 2005, 30, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Paris, J.; Schrijver, I. The digenic hypothesis unraveled: The GJB6 del(GJB6-D13S1830) mutation causes allele-specific loss of GJB2 expression in cis. Biochem. Biophys. Res. Commun. 2009, 389, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Paris, J.; Tamayo, M.L.; Gelvez, N.; Schrijver, I. Allele-specific impairment of GJB2 expression by GJB6 deletion del(GJB6-D13S1854). PLoS ONE 2011, 6, e21665. [Google Scholar] [CrossRef] [PubMed]
- Le Nabec, A.; Blotas, C.; Briset, A.; Collobert, M.; Ferec, C.; Moisan, S. 3D Chromatin Organization Involving MEIS1 Factor in the cis-Regulatory Landscape of GJB2. Int. J. Mol. Sci. 2022, 23, 6964. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas, R.; Dineiro, M.; Cifuentes, G.A.; Castillo, D.; Pruneda, P.C.; Alvarez, R.; Sanchez-Duran, N.; Capin, R.; Plasencia, A.; Viejo-Diaz, M.; et al. Comprehensive genomic diagnosis of non-syndromic and syndromic hereditary hearing loss in Spanish patients. BMC Med. Genom. 2018, 11, 58. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A.; et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Deletion | Genome Version | Centromeric Breakpoint | Telomeric Breakpoint | Reference |
|---|---|---|---|---|
| GJB6-D13S1830 | GRCh38/hg38 | chr13:20223038 | chr13:20531828 | [6] |
| GRCh37/hg19 | chr13:20797177 | chr13:21105967 | ||
| GJB6-D13S1854 | GRCh38/hg38 | chr13:20228587 | chr13:20460616 | [7] |
| GRCh37/hg19 | chr13:20802726 | chr13:21034755 | ||
| del(131 kb) | GRCh38/hg38 | chr13:20365205 | chr13:20496559 | [8] |
| GRCh37/hg19 | chr13:20939344 | chr13:21070698 | ||
| del(179 kb) | GRCh38/hg38 | chr13:20347572 | chr13:20526976 | [9] |
| GRCh37/hg19 | chr13:20921711 | chr13:21101115 | ||
| del(125kb) | GRCh38/hg38 | chr13:20398369 | chr13:20523824 | [10] |
| GRCh37/hg19 | chr13:20972508 | chr13:21097963 | ||
| del(200 kb)insATTATA | GRCh38/hg38 | chr13:20361160 | chr13:20561391 | This study |
| GRCh37/hg19 | chr13:20935299 | chr13:21135530 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cifuentes, G.A.; Diñeiro, M.; Huete, A.R.; Capín, R.; Santiago, A.; Vargas, A.A.R.; Carrero, D.; Martínez, E.L.; Aguiar, B.; Fischer, A.; et al. A Novel Recurrent 200 kb CRYL1 Deletion Underlies DFNB1A Hearing Loss in Patients from Northwestern Spain. Genes 2025, 16, 670. https://doi.org/10.3390/genes16060670
Cifuentes GA, Diñeiro M, Huete AR, Capín R, Santiago A, Vargas AAR, Carrero D, Martínez EL, Aguiar B, Fischer A, et al. A Novel Recurrent 200 kb CRYL1 Deletion Underlies DFNB1A Hearing Loss in Patients from Northwestern Spain. Genes. 2025; 16(6):670. https://doi.org/10.3390/genes16060670
Chicago/Turabian StyleCifuentes, Guadalupe A., Marta Diñeiro, Alicia R. Huete, Raquel Capín, Adrián Santiago, Alberto A. R. Vargas, Dido Carrero, Esther López Martínez, Beatriz Aguiar, Anja Fischer, and et al. 2025. "A Novel Recurrent 200 kb CRYL1 Deletion Underlies DFNB1A Hearing Loss in Patients from Northwestern Spain" Genes 16, no. 6: 670. https://doi.org/10.3390/genes16060670
APA StyleCifuentes, G. A., Diñeiro, M., Huete, A. R., Capín, R., Santiago, A., Vargas, A. A. R., Carrero, D., Martínez, E. L., Aguiar, B., Fischer, A., Rad, R., Costales, M., Cabanillas, R., & Cadiñanos, J. (2025). A Novel Recurrent 200 kb CRYL1 Deletion Underlies DFNB1A Hearing Loss in Patients from Northwestern Spain. Genes, 16(6), 670. https://doi.org/10.3390/genes16060670