
Comparative and Phylogenetic Analysis of Complete Chloroplast Genomes of Five Mangifera Species

Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. DNA Extraction, Sequencing, and Annotation
2.3. Codon Usage and RNA Editing Analysis
2.4. Identification of Repeat Sequences and Simple Sequence Repeats (SSR)
2.5. Sequence Variation Map and Variations in Inverted Repeat (IR) Sequences
2.6. Pi and Ka/Ks
2.7. Phylogenetic Analysis of Chloroplast Genomes
3. Results and Discussion
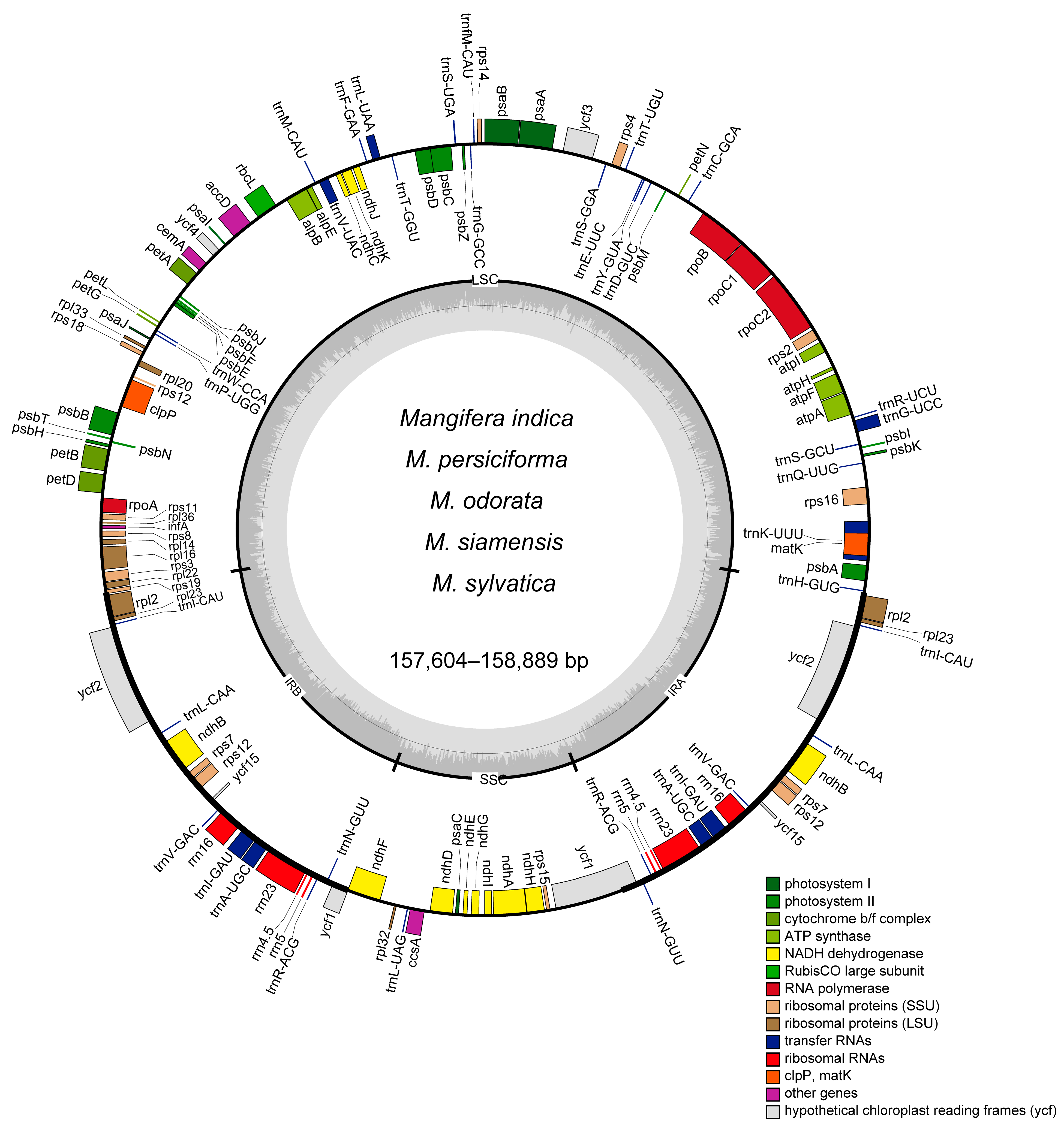
3.1. Genomic Characteristics of Chloroplast
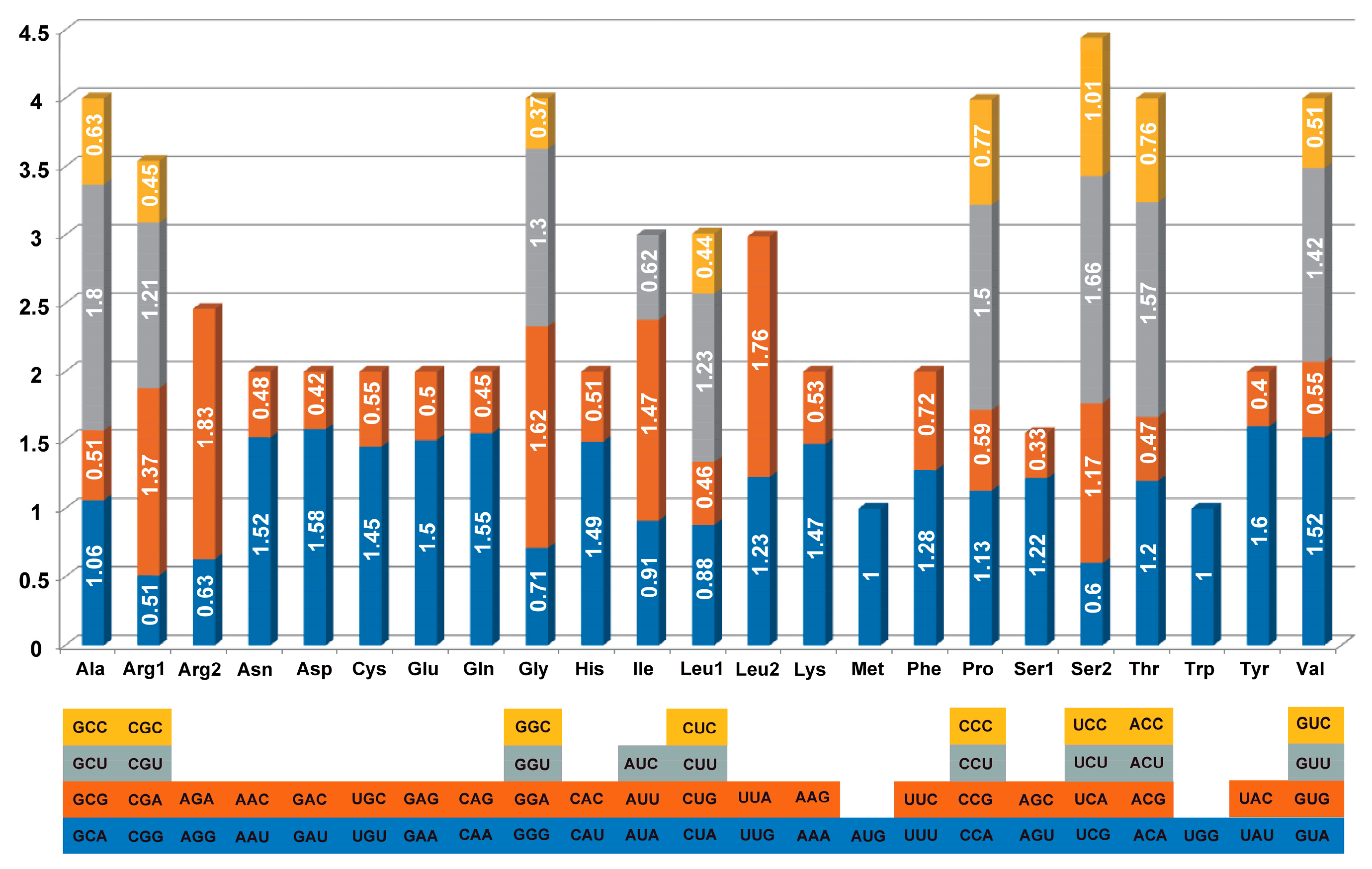
3.2. Codon Usage
3.3. RNA Editing Analysis
3.4. Non-Synonymous Substitutions (Ka) and Synonymous Substitutions (Ks) Analysis
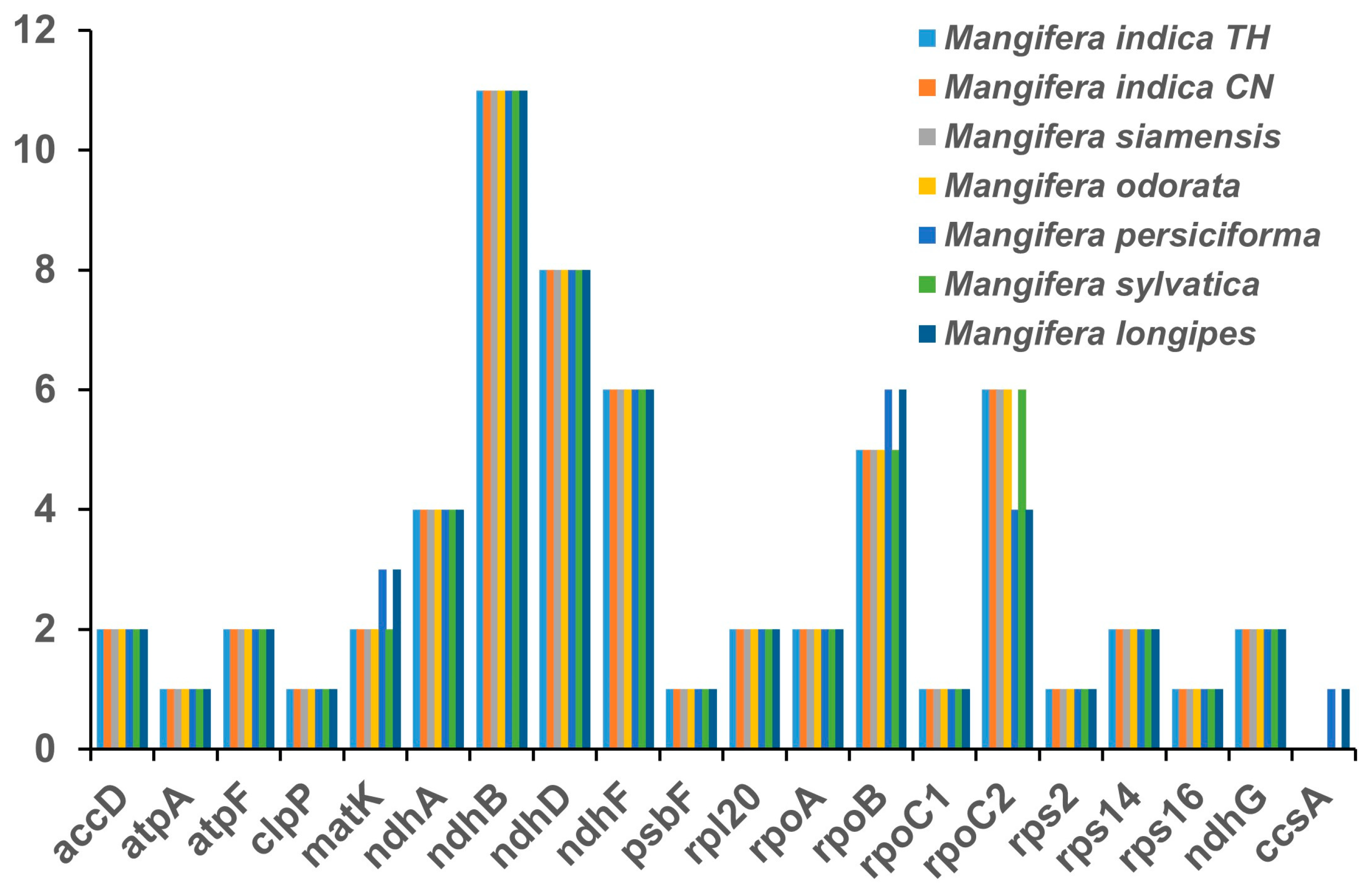
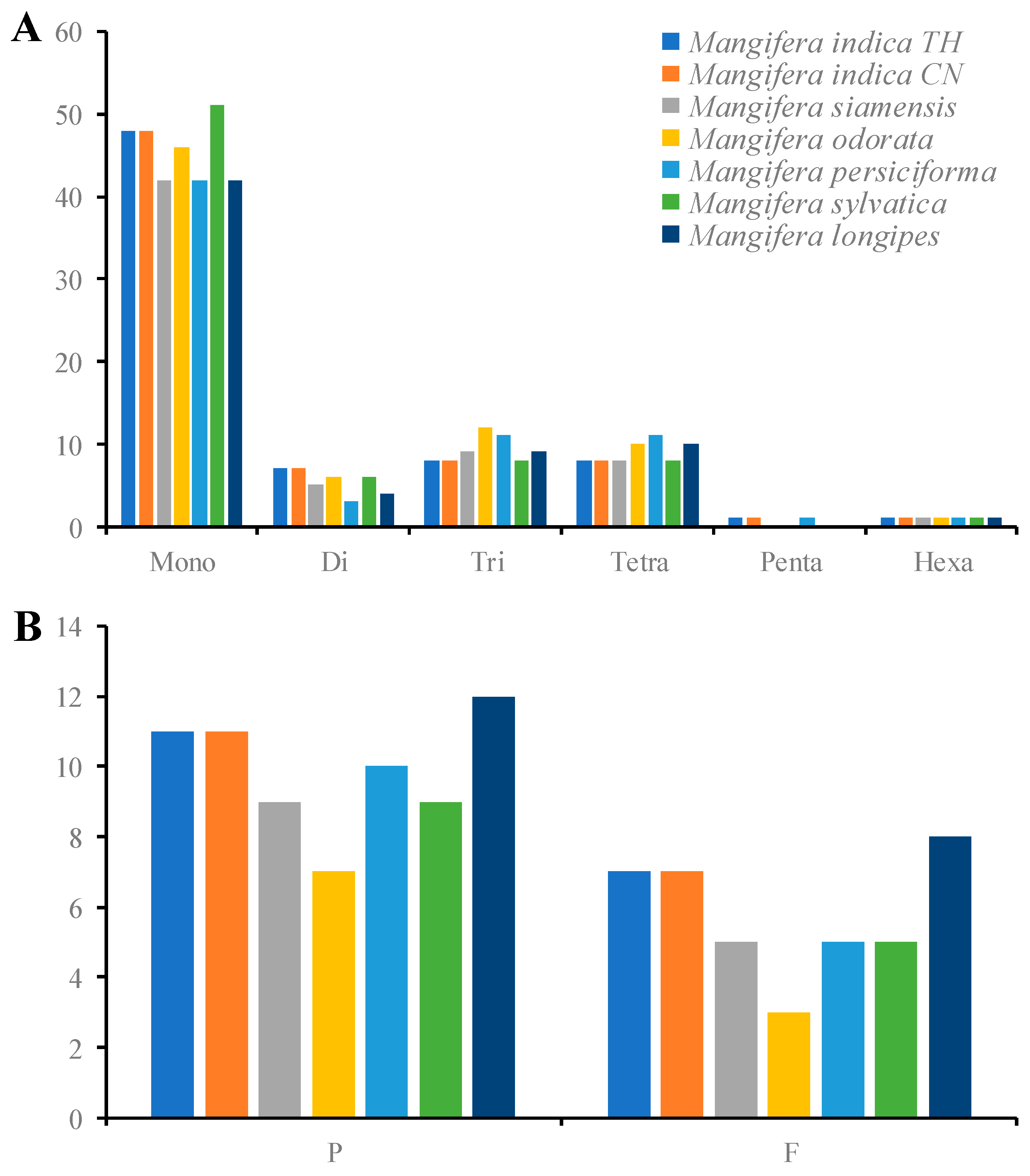
3.5. Detection of Chloroplast Repeat Sequences and SSRs
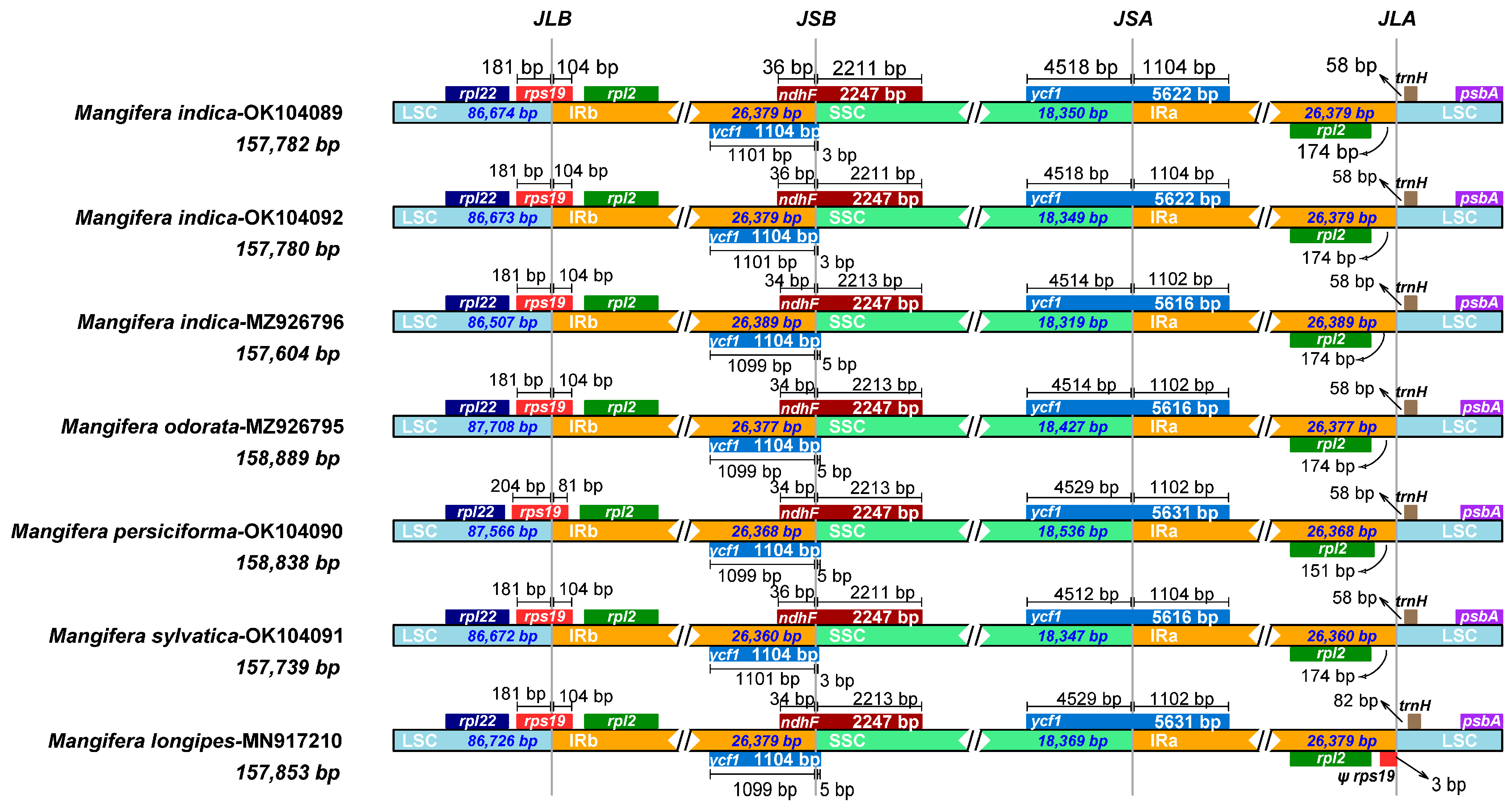
3.6. IR Contraction and Expansion
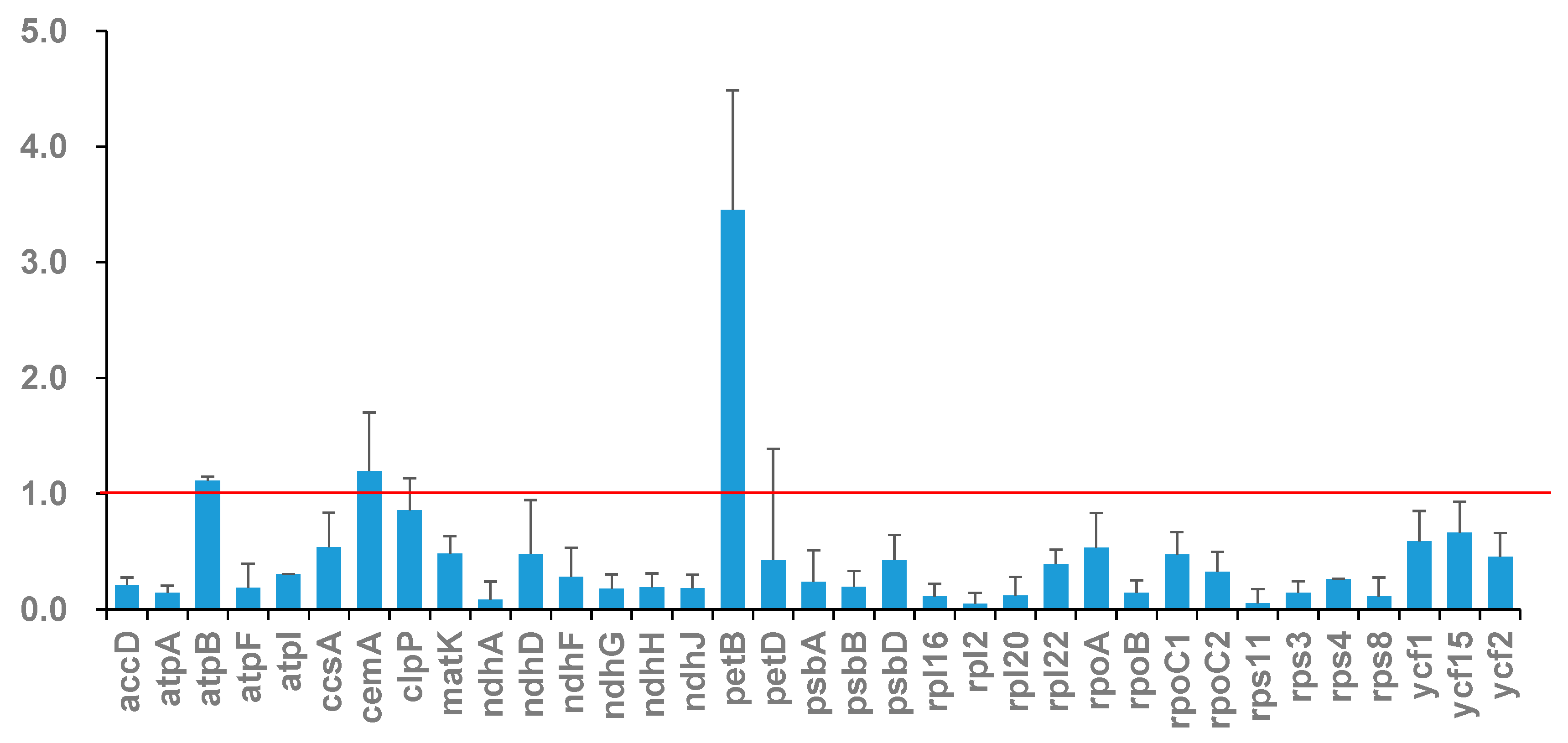
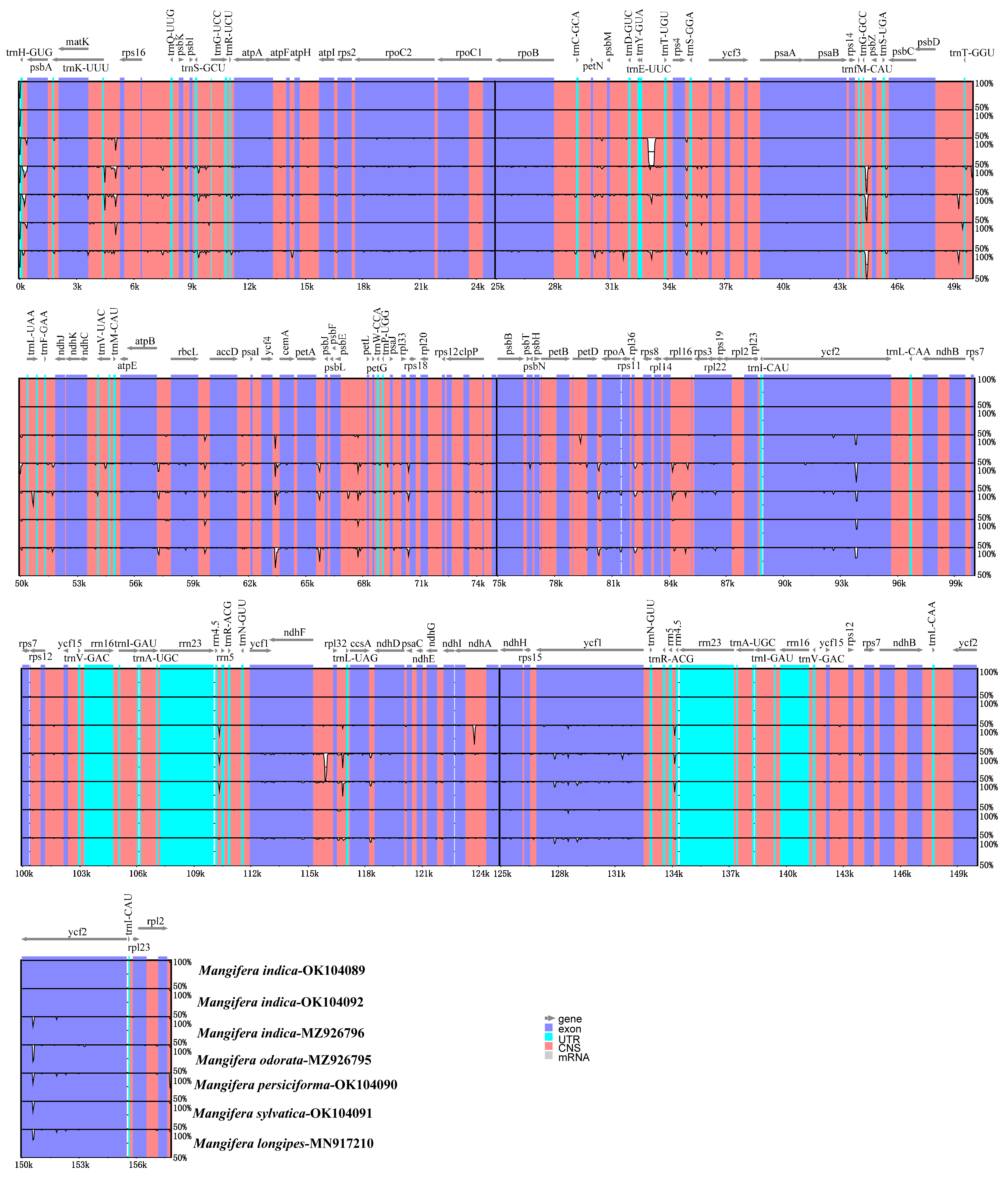
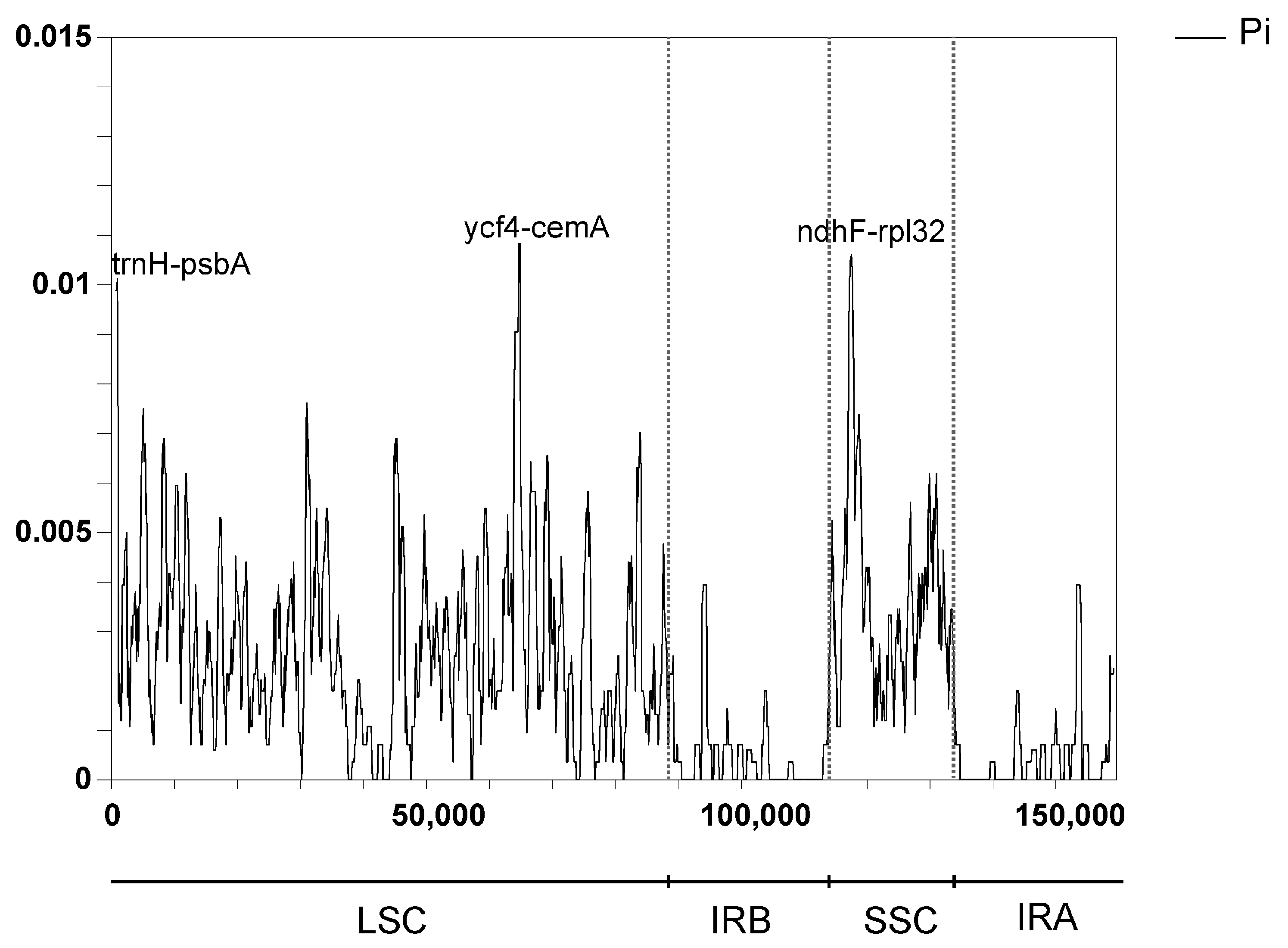
3.7. Sequence Divergence Analysis
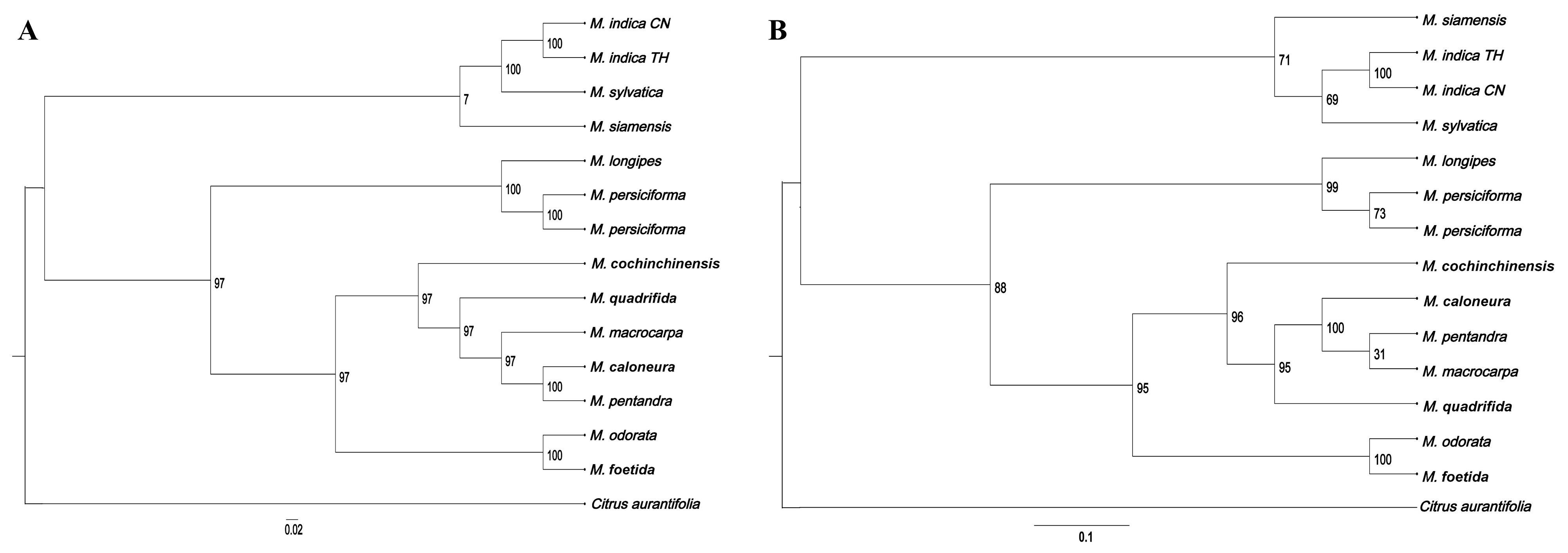
3.8. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| cp | chloroplast |
| LSC | large single copy |
| SSC | small single copy |
| IR | inverted repeat |
| RSCU | relative synonymous codon usage |
| SSR | simple sequence repeats |
| Pi | nucleotide variability |
| PCGs | protein-coding genes |
| Ka | non-synonymous substitutions |
| Ks | synonymous substitutions |
| RFLP | restriction fragment length polymorphism |
References
- Bajpai, A.; Muthukumar, M.; Ahmad, I.; Ravishankar, K.; Parthasarthy, V.; Sthapit, B.; Rao, R.; Verma, J.; Rajan, S. Molecular and morphological diversity in locally grown non-commercial (heirloom) mango varieties of North India. J. Environ. Biol. 2016, 37, 221. [Google Scholar]
- Iquebal, M.; Jaiswal, S.; Mahato, A.K.; Jayaswal, P.K.; Angadi, U.; Kumar, N.; Sharma, N.; Singh, A.K.; Srivastav, M.; Prakash, J. MiSNPDb: A web-based genomic resources of tropical ecology fruit mango (Mangifera indica L.) for phylogeography and varietal differentiation. Sci. Rep. 2017, 7, 14968. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Srivastav, M.; Rymbai, H.; Chaudhary, R.; Singh, A.; Dubey, A.; Lal, K. Pollen–pistil interaction studies in mango (Mangifera indica L.) cultivars. Sci. Hortic. 2013, 160, 213–221. [Google Scholar] [CrossRef]
- Lora, J.; Hormaza, J.I. Pollen wall development in mango (Mangifera indica L., Anacardiaceae). Plant Reprod. 2018, 31, 385–397. [Google Scholar] [CrossRef]
- Sherman, A.; Rubinstein, M.; Eshed, R.; Benita, M.; Ish-Shalom, M.; Sharabi-Schwager, M.; Rozen, A.; Saada, D.; Cohen, Y.; Ophir, R. Mango (Mangifera indica L.) germplasm diversity based on single nucleotide polymorphisms derived from the transcriptome. BMC Plant Biol. 2015, 15, 277. [Google Scholar] [CrossRef]
- Ravishankar, K.; Dinesh, M.; Mani, B.; Padmakar, B.; Vasugi, C. Assessment of genetic diversity of mango (Mangifera indica L.) cultivars from Indian Peninsula using Sequence Tagged Microsatellite Site (STMS) markers. In Proceedings of the IX International Mango Symposium 992, Sanya, China, 8–12 April 2010; pp. 269–276. [Google Scholar]
- Mansour, H.; Mekki, L.E.; Hussein, M.A. Assessment of genetic diversity and relationships among Egyptian mango (Mangifera indica L.) cultivers grown in Suez Canal and Sinai region using RAPD markers. Pak. J. Biol. Sci. 2014, 17, 56–61. [Google Scholar] [CrossRef]
- Nishiyama, K.; Choi, Y.A.; Honsho, C.; Eiadthong, W.; Yonemori, K. Application of genomic in situ hybridization for phylogenetic study between Mangifera indica L. and eight wild species of Mangifera. Sci. Hortic. 2006, 110, 114–117. [Google Scholar] [CrossRef]
- Sankaran, M.; Dinesh, M.; Chaitra, N.; Ravishankar, K. Morphological, cytological, palynological and molecular characterization of certain Mangifera species. Curr. Sci. 2018, 115, 1379–1386. [Google Scholar] [CrossRef]
- Eiadthong, W.; Yonemori, K.; Sugiura, A.; Utsunomiya, N.; Subhadrabandhu, S. Analysis of phylogenetic relationships in Mangifera by restriction site analysis of an amplified region of cpDNA. Sci. Hortic. 1999, 80, 145–155. [Google Scholar] [CrossRef]
- Fitmawati, F.; Harahap, S.P.; Sofiyanti, N. Phylogenetic analysis of mango (Mangifera) in Northern Sumatra based on gene sequences of cpDNA trnL-F intergenic spacer. Biodiversitas J. Biol. Divers. 2017, 18, 715–719. [Google Scholar] [CrossRef]
- Fitmawati, F.; Hayati, I.; Sofiyanti, N. Using ITS as a molecular marker for Mangifera species identification in Central Sumatra. Biodiversitas J. Biol. Divers. 2016, 17, 653–656. [Google Scholar]
- Yonemori, K.; Honsho, C.; Kanzaki, S.; Eiadthong, W.; Sugiura, A. Phylogenetic relationships of Mangifera species revealed by ITS sequences of nuclear ribosomal DNA and a possibility of their hybrid origin. Plant Syst. Evol. 2002, 231, 59–75. [Google Scholar] [CrossRef]
- Litz, R.E. The Mango: Botany, Production and Uses; CABI: Wallingford, UK, 2009. [Google Scholar]
- Howe, C.J.; Barbrook, A.C.; Koumandou, V.L.; Nisbet, R.E.R.; Symington, H.A.; Wightman, T.F. Evolution of the chloroplast genome. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2003, 358, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Chen, Z.; Shi, W.; Han, W.; Feng, Q.; Shi, C.; Engel, M.S.; Wang, S. Comparative analysis of complete chloroplast genomes of nine species of Litsea (Lauraceae): Hypervariable regions, positive selection, and phylogenetic relationships. Genes 2022, 13, 1550. [Google Scholar] [CrossRef]
- Song, W.; Ji, C.; Chen, Z.; Cai, H.; Wu, X.; Shi, C.; Wang, S. Comparative analysis the complete chloroplast genomes of nine Musa species: Genomic features, comparative analysis, and phylogenetic implications. Front. Plant Sci. 2022, 13, 832884. [Google Scholar] [CrossRef]
- Tang, J.; Zou, R.; Chen, T.; Pan, L.; Zhu, S.; Ding, T.; Chai, S.; Wei, X. Comparative Analysis of the Complete Chloroplast Genomes of Six Endangered Cycas Species: Genomic Features, Comparative Analysis, and Phylogenetic Implications. Forests 2023, 14, 2069. [Google Scholar] [CrossRef]
- Tang, J.; Zou, R.; Huang, K.; Gao, L.; Tang, F.; Ding, T.; Jiang, Y.; Wei, X. Comparative and Phylogenetic Analyses of the Complete Chloroplast Genomes of Four Ottelia Species. Horticulturae 2024, 10, 603. [Google Scholar] [CrossRef]
- Wang, R.-J.; Cheng, C.-L.; Chang, C.-C.; Wu, C.-L.; Su, T.-M.; Chaw, S.-M. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 2008, 8, 36. [Google Scholar] [CrossRef]
- Cui, Y.; Nie, L.; Sun, W.; Xu, Z.; Wang, Y.; Yu, J.; Song, J.; Yao, H. Comparative and phylogenetic analyses of ginger (Zingiber officinale) in the family Zingiberaceae based on the complete chloroplast genome. Plants 2019, 8, 283. [Google Scholar] [CrossRef]
- Chen, H.; Deng, L.; Jiang, Y.; Lu, P.; Yu, J. RNA editing sites exist in protein-coding genes in the chloroplast genome of Cycas taitungensis. J. Integr. Plant Biol. 2011, 53, 961–970. [Google Scholar] [CrossRef]
- Niu, Y.; Gao, C.; Liu, J. Comparative analysis of the complete plastid genomes of Mangifera species and gene transfer between plastid and mitochondrial genomes. PeerJ 2021, 9, e10774. [Google Scholar] [CrossRef] [PubMed]
- Wijesundara, U.K.; Furtado, A.; Dillon, N.L.; Masouleh, A.K.; Henry, R.J. Phylogenetic relationships in the genus Mangifera based on whole chloroplast genome and nuclear genome sequences. Trop. Plants 2024, 3, e034. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Qu, X.-J.; Moore, M.J.; Li, D.-Z.; Yi, T.-S. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes. Plant Methods 2019, 15, 50. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Team, U. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Dobrogojski, J.; Adamiec, M.; Luciński, R. The chloroplast genome: A review. Acta Physiol. Plant. 2020, 42, 98. [Google Scholar] [CrossRef]
- Zhang, Y.; Ou, K.-W.; Huang, G.-D.; Lu, Y.-F.; Yang, G.-Q.; Pang, X.-H. The complete chloroplast genome sequence of Mangifera sylvatica Roxb.(Anacardiaceae) and its phylogenetic analysis. Mitochondrial DNA Part B 2020, 5, 738–739. [Google Scholar] [CrossRef]
- Asaf, S.; Khan, A.L.; Khan, A.R.; Waqas, M.; Kang, S.-M.; Khan, M.A.; Lee, S.-M.; Lee, I.-J. Complete chloroplast genome of Nicotiana otophora and its comparison with related species. Front. Plant Sci. 2016, 7, 843. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, N.; Wu, S.; Jiang, C.; Xie, L.; Yang, F.; Yu, Z. A comparative analysis of the chloroplast genomes of three Lonicera medicinal plants. Genes 2023, 14, 548. [Google Scholar] [CrossRef]
- Mehmood, F.; Shahzadi, I.; Waseem, S.; Mirza, B.; Ahmed, I.; Waheed, M.T. Chloroplast genome of Hibiscus rosa-sinensis (Malvaceae): Comparative analyses and identification of mutational hotspots. Genomics 2020, 112, 581–591. [Google Scholar]
- Chen, J.; Hao, Z.; Xu, H.; Yang, L.; Liu, G.; Sheng, Y.; Zheng, C.; Zheng, W.; Cheng, T.; Shi, J. The complete chloroplast genome sequence of the relict woody plant Metasequoia glyptostroboides Hu et Cheng. Front. Plant Sci. 2015, 6, 447. [Google Scholar] [CrossRef]
- Raman, G.; Park, S. Analysis of the complete chloroplast genome of a medicinal plant, Dianthus superbus var. Longicalyncinus, from a comparative genomics perspective. PLoS ONE 2015, 10, e0141329. [Google Scholar]
- Mower, J.P. PREP-Mt: Predictive RNA editor for plant mitochondrial genes. BMC Bioinform. 2005, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Fay, J.C.; Wu, C.-I. Sequence divergence, functional constraint, and selection in protein evolution. Annu. Rev. Genom. Hum. Genet. 2003, 4, 213–235. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Zhao, X.-Q.; Wang, J.; Wong, G.K.-S.; Yu, J. KaKs_Calculator: Calculating Ka and Ks through model selection and model averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Fu, P.-C.; Zhang, Y.-Z.; Geng, H.-M.; Chen, S.-L. The complete chloroplast genome sequence of Gentiana lawrencei var. farreri (Gentianaceae) and comparative analysis with its congeneric species. PeerJ 2016, 4, e2540. [Google Scholar] [PubMed]
- Lin, Y.; Chen, Y.; Zhao, Y.; Wu, W.; Yang, C.; Zheng, Y.; Huang, M. Comparative Analysis of Complete Chloroplast Genomes and Phylogenetic Relationships in Medicinally Important Pantropical Genus Bauhinia ss (Leguminosae) from Southern Africa and Eastern Asia. Int. J. Mol. Sci. 2025, 26, 397. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, Q.; Zhang, C.; Huang, H.; He, H.; Wang, M.; Li, M.; Huang, Z.; Tang, Y.; Chen, Q. Sequencing and analysis of complete chloroplast genomes provide insight into the evolution and phylogeny of Chinese kale (Brassica oleracea var. alboglabra). Int. J. Mol. Sci. 2023, 24, 10287. [Google Scholar] [CrossRef]
- Heyno, E.; Ermakova, M.; Lopez-Calcagno, P.E.; Woodford, R.; Brown, K.L.; Matthews, J.S.; Osmond, B.; Raines, C.A.; von Caemmerer, S. Rieske FeS overexpression in tobacco provides increased abundance and activity of cytochrome b 6 f. Physiol. Plant. 2022, 174, e13803. [Google Scholar] [CrossRef]
- Liang, C.; Wang, L.; Lei, J.; Duan, B.; Ma, W.; Xiao, S.; Qi, H.; Wang, Z.; Liu, Y.; Shen, X. A comparative analysis of the chloroplast genomes of four Salvia medicinal plants. Engineering 2019, 5, 907–915. [Google Scholar] [CrossRef]
- Yaradua, S.S.; Alzahrani, D.A.; Albokhary, E.J.; Abba, A.; Bello, A. Complete chloroplast genome sequence of Justicia flava: Genome comparative analysis and phylogenetic relationships among Acanthaceae. BioMed Res. Int. 2019, 2019, 4370258. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, T.; Duan, D.; Yang, J.; Feng, L.; Zhao, G. Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 2016, 7, 959. [Google Scholar] [CrossRef]
- Wang, J.; Qian, J.; Jiang, Y.; Chen, X.; Zheng, B.; Chen, S.; Yang, F.; Xu, Z.; Duan, B. Comparative analysis of chloroplast genome and new insights into phylogenetic relationships of Polygonatum and tribe Polygonateae. Front. Plant Sci. 2022, 13, 882189. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dong, Y.; Liu, Y.; Yu, X.; Yang, M.; Huang, Y. Comparative analyses of Euonymus chloroplast genomes: Genetic structure, screening for loci with suitable polymorphism, positive selection genes, and phylogenetic relationships within Celastrineae. Front. Plant Sci. 2021, 11, 593984. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Huang, T.; Zhou, Q.; Sheng, Q.; Zhu, Z. Complete chloroplast genomes and phylogenetic relationships of Bougainvillea spectabilis and Bougainvillea glabra (Nyctaginaceae). Int. J. Mol. Sci. 2023, 24, 13044. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.; Xue, Q.; Wang, H.; Xie, X.; Zhu, S.; Liu, W.; Ding, X. Mutational biases and GC-biased gene conversion affect GC content in the plastomes of Dendrobium genus. Int. J. Mol. Sci. 2017, 18, 2307. [Google Scholar] [CrossRef]
- Qian, J.; Song, J.; Gao, H.; Zhu, Y.; Xu, J.; Pang, X.; Yao, H.; Sun, C.; Li, X.; Li, C. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PLoS ONE 2013, 8, e57607. [Google Scholar] [CrossRef]
- Wang, P.; Luo, Y.; Huang, J.; Gao, S.; Zhu, G.; Dang, Z.; Gai, J.; Yang, M.; Zhu, M.; Zhang, H. The genome evolution and domestication of tropical fruit mango. Genome Biol. 2020, 21, 60. [Google Scholar] [CrossRef]
- Kelchner, S.A. The evolution of non-coding chloroplast DNA and its application in plant systematics. Ann. Mo. Bot. Gard. 2000, 87, 482–498. [Google Scholar] [CrossRef]
- Yadav, I. Occurrence of races of Mangifera species. Indian J. Hortic. 1999, 56, 96–103. [Google Scholar]
- Xia, C.; Wang, M.; Guan, Y.; Li, J. Comparative analysis of the chloroplast genome for Aconitum species: Genome structure and phylogenetic relationships. Front. Genet. 2022, 13, 878182. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | M. indica | M. indica | M. siamensis | M. odorata | M. persiciforma | M. sylvatica |
|---|---|---|---|---|---|---|
| Accession number | OK104089 | OK104092 | MZ926796 | MZ926795 | OK104090 | OK104091 |
| Size | 157,782 | 157,780 | 157,604 | 158,889 | 158,838 | 157,739 |
| GC content | 37.89% | 37.89% | 37.90% | 37.81% | 37.82% | 37.89% |
| IR size | 26,379 | 26,379 | 26,389 | 26,377 | 26,368 | 26,360 |
| LSC size | 86,674 | 86,673 | 86,507 | 87,708 | 87,566 | 86,672 |
| SSC size | 18,350 | 18,349 | 18,319 | 18,427 | 18,536 | 18,347 |
| Genes | 134 | 133 | 133 | 133 | 133 | 133 |
| PCGs | 89 | 88 | 88 | 88 | 88 | 88 |
| tRNA | 37 | 37 | 37 | 37 | 37 | 37 |
| rRNA | 8 | 8 | 8 | 8 | 8 | 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, Y.; Yang, X.; Luo, S.; Huang, G.; Zhang, Y.; Zhao, Y.; Li, R.; Guo, L.; Ran, M.; Gao, A.; et al. Comparative and Phylogenetic Analysis of Complete Chloroplast Genomes of Five Mangifera Species. Genes 2025, 16, 666. https://doi.org/10.3390/genes16060666
Tang Y, Yang X, Luo S, Huang G, Zhang Y, Zhao Y, Li R, Guo L, Ran M, Gao A, et al. Comparative and Phylogenetic Analysis of Complete Chloroplast Genomes of Five Mangifera Species. Genes. 2025; 16(6):666. https://doi.org/10.3390/genes16060666
Chicago/Turabian StyleTang, Yujuan, Xiangyan Yang, Shixing Luo, Guodi Huang, Yu Zhang, Ying Zhao, Riwang Li, Limei Guo, Mengyang Ran, Aiping Gao, and et al. 2025. "Comparative and Phylogenetic Analysis of Complete Chloroplast Genomes of Five Mangifera Species" Genes 16, no. 6: 666. https://doi.org/10.3390/genes16060666
APA StyleTang, Y., Yang, X., Luo, S., Huang, G., Zhang, Y., Zhao, Y., Li, R., Guo, L., Ran, M., Gao, A., & Huang, J. (2025). Comparative and Phylogenetic Analysis of Complete Chloroplast Genomes of Five Mangifera Species. Genes, 16(6), 666. https://doi.org/10.3390/genes16060666