
Retinal Changes in Early-Onset cblC Methylmalonic Acidemia Identified Through Expanded Newborn Screening: Highlights from a Case Study and Literature Review

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient
2.2. Newborn Screening and Confirmatory Testing
2.3. Molecular Analysis
2.4. Visual and Ophthalmologic Testing
2.5. Literature Review
3. Results
3.1. Clinical Report
3.2. Biochemical Features
3.3. Genetic Findings
3.4. Digenic Methylmalonic Acidemia and Homocystinuria, cblC Type (epi-cblC)
3.5. Literature Mining
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| cblC | Methylmalonic acidemia combined with homocystinuria |
| BEM | Bull’s Eye Maculopathy |
| NBS | Newborn Screening |
| DBS | Dried Blood Spot |
| OCT | Optical Coherence Tomography |
| ERG | Electroretinography |
| C3 | Propionylcarnitine |
| MMA | Methylmalonic acid |
| Hcy | Homocysteine |
| Met | Methionine |
| OHCbl | Hydroxocobalamin |
| OPL | Outer Plexiform Layer |
| ONL | Outer Nuclear Layer |
| FF | Fix and Follow |
| CSM | Central, Steady, Maintained |
References
- Ruoppolo, M.; Malvagia, S.; Boenzi, S.; Carducci, C.; Dionisi-Vici, C.; Teofoli, F.; Burlina, A.; Angeloni, A.; Aronica, T.; Bordugo, A.; et al. Expanded Newborn Screening in Italy Using Tandem Mass Spectrometry: Two Years of National Experience. Int. J. Neonatal Screen. 2022, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Huemer, M.; Diodato, D.; Schwahn, B.; Schiff, M.; Bandeira, A.; Benoist, J.F.; Burlina, A.; Cerone, R.; Couce, M.L.; Garcia-Cazorla, A.; et al. Guidelines for diagnosis and management of the cobalamin-related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR deficiency. J. Inherit. Metab. Dis. 2017, 40, 21–48. [Google Scholar] [CrossRef] [PubMed]
- Guéant, J.L.; Chéry, C.; Oussalah, A.; Nadaf, J.; Coelho, D.; Josse, T.; Flayac, J.; Robert, A.; Koscinski, I.; Gastin, I.; et al. APRDX1 mutant allele causes a MMACHC secondary epimutation in cblC patients. Nat. Commun. 2018, 9, 67, Erratum in Nat. Commun. 2018, 9, 554. [Google Scholar] [CrossRef]
- Weisfeld-Adams, J.D.; McCourt, E.A.; Diaz, G.A.; Oliver, S.C. Ocular disease in the cobalamin C defect: A review of the literature and a suggested framework for clinical surveillance. Mol. Genet. Metab. 2015, 114, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Feresin, A.; Spedicati, B.; Zampieri, S.; Morgan, A.; Magnolato, A.; Tesser, A.; Tommasini, A.; Bonati, M.T.; Girotto, G.; Faletra, F. Does It Run in Your Family? Inherited Truncating PSMD12 Variants Broaden the Phenotypic Spectrum of Stankiewicz-Isidor Syndrome. Am. J. Med. Genet. Part A 2024, 197, e63953. [Google Scholar] [CrossRef]
- Daneshvar, R.; Ehsaei, A.; Moghadas Sharif, N.; Pato, Z. Comparison of Visual Field Measurements in Glaucomatous Eyes using Oculus and Metrovision Perimeters. J. Curr. Ophthalmol. 2023, 35, 17–22. [Google Scholar] [CrossRef]
- Maines, E.; Morandi, G.; Gugelmo, G.; Ion-Popa, F.; Campostrini, N.; Pasini, A.; Vincenzi, M.; Teofoli, F.; Camilot, M.; Bordugo, A. Vitamin B12 Administration by Subcutaneous Catheter Device in a Cobalamin A (cblA) Patient. JIMD Rep. 2017, 35, 29–31. [Google Scholar]
- Gizicki, R.; Robert, M.C.; Gómez-López, L.; Orquin, J.; Decarie, J.C.; Mitchell, G.A.; Roy, M.S.; Ospina, L.H. Long-term visual outcome of methylmalonic aciduria and homocystinuria, cobalamin C type. Ophthalmology 2014, 121, 381–386. [Google Scholar] [CrossRef]
- Cavicchi, C.; Oussalah, A.; Falliano, S.; Ferri, L.; Gozzini, A.; Gasperini, S.; Motta, S.; Rigoldi, M.; Parenti, G.; Tummolo, A.; et al. PRDX1 gene-related epi-cblC disease is a common type of inborn error of cobalamin metabolism with mono- or bi-allelic MMACHC epimutations. Clin. Epigenet. 2021, 13, 137. [Google Scholar] [CrossRef]
- Ku, C.A.; Ng, J.K.; Karr, D.J.; Reznick, L.; Harding, C.O.; Weleber, R.G.; Pennesi, M.E. Spectrum of ocular manifestations in cobalamin C and cobalamin A types of methylmalonic acidemia. Ophthalmic Genet. 2016, 37, 404–414. [Google Scholar] [CrossRef]
- Fuchs, L.R.; Robert, M.; Ingster-Moati, I.; Couette, L.; Dufier, J.L.; de Lonlay, P.; Brodie, S.E. Ocular manifestations of cobalamin C type methylmalonic aciduria with homocystinuria. J. AAPOS 2012, 16, 370–375. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Mo, R.; Shen, M.; Kang, L.; Song, J.; Liu, Y.; Chen, Z.; Zhang, H.; Yao, H.; Liu, Y.; et al. Variable phenotypes and outcomes associated with the MMACHC c.609G>A homologous mutation: Long term follow-up in a large cohort of cases. Orphanet J. Rare Dis. 2020, 15, 200. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Wu, S.; Shuai, R.; Yu, Y.; Qiu, W.; Wei, H.; Yang, C.; Xu, P.; Zou, H.; Feng, J.; et al. The Follow-Up of Chinese Patients in cblC Type Methylmalonic Acidemia Identified Through Expanded Newborn Screening. Front. Genet. 2022, 13, 805599. [Google Scholar] [CrossRef]
- Carrillo-Carrasco, N.; Venditti, C.P. Combined methylmalonic acidemia and homocystinuria, cblC type. II. Complications, pathophysiology, and outcomes. J. Inherit. Metab. Dis. 2012, 35, 103–114. [Google Scholar] [CrossRef]
- Collison, F.T.; Xie, Y.A.; Gambin, T.; Jhangiani, S.; Muzny, D.; Gibbs, R.; Lupski, J.R.; Fishman, G.A.; Allikmets, R. Whole Exome Sequencing Identifies an Adult-Onset Case of Methylmalonic Aciduria and Homocystinuria Type C (cblC) with Non-Syndromic Bull’s Eye Maculopathy. Ophthalmic Genet. 2015, 36, 270–275. [Google Scholar] [CrossRef]
- Kiessling, E.; Nötzli, S.; Todorova, V.; Forny, M.; Baumgartner, M.R.; Samardzija, M.; Krijt, J.; Kožich, V.; Grimm, C.; Froese, D.S. Absence of MMACHC in peripheral retinal cells does not lead to an ocular phenotype in mice. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166201. [Google Scholar] [CrossRef] [PubMed]
- Aleman, T.S.; Brodie, F.; Garvin, C.; Gewaily, D.Y.; Ficicioglu, C.H.; Mills, M.D.; Forbes, B.J.; Maguire, A.M.; Davidson, S.L. Retinal Structure in Cobalamin C Disease: Mechanistic and Therapeutic Implications. Ophthalmic Genet. 2015, 36, 339–348. [Google Scholar] [CrossRef]
- Pastore, A.; Martinelli, D.; Piemonte, F.; Tozzi, G.; Boenzi, S.; Di Giovamberardino, G.; Petrillo, S.; Bertini, E.; Dionisi-Vici, C. Glutathione metabolism in cobalamin deficiency type C (cblC). J. Inherit. Metab. Dis. 2014, 37, 125–129. [Google Scholar] [CrossRef]
- Sun, M.; Dai, Y. Late-onset cobalamin C deficiency type in adult with cognitive and behavioral disturbances and significant cortical atrophy and cerebellar damage in the MRI: A case report. Front. Neurol. 2023, 14, 1308289. [Google Scholar] [CrossRef]
- Paz, D.; Pinales, B.E.; Castellanos, B.S.; Perez, I.; Gil, C.B.; Madrigal, L.J.; Reyes-Nava, N.G.; Castro, V.L.; Sloan, J.L.; Quintana, A.M. Abnormal chondrocyte development in a zebrafish model of cblC syndrome restored by an MMACHC cobalamin binding mutant. Differentiation 2023, 131, 74–81. [Google Scholar] [CrossRef]
- Martinelli, D.; Dotta, A.; Massella, L.; Picca, S.; Di Pede, A.; Boenzi, S.; Aiello, C.; Dionisi-Vici, C. Cobalamin C defect presenting as severe neonatal hyperammonemia. Eur. J. Pediatr. 2011, 170, 887–890. [Google Scholar] [CrossRef] [PubMed]
- Grant, L.W.; McCandless, S.E.; Traboulsi, E.I. Maculopathy Due to Cobalamin C (cb1C) Disease in an Amish Child. J. Pediatr. Ophthalmol. Strabismus 2010, 47, 1–3. [Google Scholar] [CrossRef]
- Weisfeld-Adams, J.D.; Bender, H.A.; Miley-Åkerstedt, A.; Frempong, T.; Schrager, N.L.; Patel, K.; Naidich, T.P.; Stein, V.; Spat, J.; Towns, S.; et al. Neurologic and neurodevelopmental phenotypes in young children with early-treated combined methylmalonic acidemia and homocystinuria, cobalamin C type. Mol. Genet. Metab. 2013, 110, 241–247. [Google Scholar] [CrossRef]
- Schimel, A.M.; Mets, M.B. The natural history of retinal degeneration in association with cobalamin C (cbl C) disease. Ophthalmic Genet. 2006, 27, 9–14. [Google Scholar] [CrossRef]
- Gaillard, M.C.; Matthieu, J.M.; Borruat, F.X. Retinal dysfunction in combined methylmalonic aciduria and homocystinuria (Cblc) disease: A spectrum of disorders. Klin. Monatsblätter Augenheilkd. 2008, 225, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Froese, D.S.; Fowler, B.; Baumgartner, M.R. Vitamin B12, folate, and the methionine remethylation cycle-biochemistry, pathways, and regulation. J. Inherit. Metab. Dis. 2019, 42, 673–685. [Google Scholar] [CrossRef]
- Zeviani, M.; Carelli, V. Mitochondrial Retinopathies. Int. J. Mol. Sci. 2021, 23, 210. [Google Scholar] [CrossRef] [PubMed]
- Rajappa, M.; Goyal, A.; Kaur, J. Inherited metabolic disorders involving the eye: A clinico-biochemical perspective. Eye 2010, 24, 507–518. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Wolf, J.; Boneva, S.; Schlecht, A.; Lapp, T.; Auw-Haedrich, C.; Lagrèze, W.; Agostini, H.; Reinhard, T.; Schlunck, G.; Lange, C. The Human Eye Transcriptome Atlas: A searchable comparative transcriptome database for healthy and diseased human eye tissue. Genomics 2022, 114, 110286. [Google Scholar] [CrossRef]
- Traboulsi, E.I.; Silva, J.C.; Geraghty, M.T.; Maumenee, I.H.; Valle, D.; Green, W.R. Ocular histopathologic characteristics of cobalamin C type vitamin B12 defect with methylmalonic aciduria and homocystinuria. Am. J. Ophthalmol. 1992, 113, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Suppiej, A.; Marino, S.; Reffo, M.E.; Maritan, V.; Vitaliti, G.; Mailo, J.; Falsaperla, R. Early onset retinal dystrophies: Clinical clues to diagnosis for pediatricians. Ital. J. Pediatr. 2019, 45, 168. [Google Scholar] [CrossRef]
- Gerth, C.; Morel, C.F.; Feigenbaum, A.; Levin, A.V. Ocular phenotype in patients with methylmalonic aciduria and homocystinuria, cobalamin C type. J. AAPOS 2008, 12, 591–596, Erratum in J. AAPOS 2009, 13, 223. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, S.; Brezzi, B.; Bracciamà, V.; Barreca, A.; Nozza, P.; Vaisitti, T.; Amoroso, A.; Deaglio, S.; Manganaro, M.; Porta, F.; et al. Adult-onset CblC deficiency: A challenging diagnosis involving different adult clinical specialists. Orphanet J. Rare Dis. 2022, 17, 33. [Google Scholar] [CrossRef]
- Kinoshita, Y.; Nogami, K.; Jomura, R.; Akanuma, S.I.; Abe, H.; Inouye, M.; Kubo, Y.; Hosoya, K.I. Investigation of Receptor-Mediated Cyanocobalamin (Vitamin B12) Transport across the Inner Blood-Retinal Barrier Using Fluorescence-Labeled Cyanocobalamin. Mol. Pharm. 2018, 15, 3583–3594. [Google Scholar] [CrossRef]
- Luder, A.S.; Tanner, S.M.; de la Chapelle, A.; Walter, J.H. Amnionless (AMN) mutations in Imerslund-Gräsbeck syndrome may be associated with disturbed vitamin B12 transport into the CNS. J. Inherit. Metab. Dis. 2008, 31 (Suppl. S3), 493–496. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, G.; Greco, B.; Cairoli, S.; Catesini, G.; Lepri, F.R.; Orazi, L.; Mallardi, M.; Martinelli, D.; Ricci, D.; Simeoli, R.; et al. Improved biochemical and neurodevelopmental profiles with high-dose hydroxocobalamin therapy in cobalamin C defect. J. Inherit. Metab. Dis. 2025, 48, e12787. [Google Scholar] [CrossRef]
- Scalais, E.; Geron, C.; Pierron, C.; Cardillo, S.; Schlesser, V.; Mataigne, F.; Borde, P.; Regal, L. Would, early, versus late hydroxocobalamin dose intensification treatment, prevent cognitive decline, macular degeneration and ocular disease, in 5 patients with early-onset cblC deficiency? Mol. Genet. Metab. 2023, 140, 107681. [Google Scholar] [CrossRef]
- Venditti, C.; Sloan, J.; Zein, W.; Thurm, A.; Hall, C.; Van Ryzin, C.; Ferry, S.; Gebremariam, A.; Myles, J.; Huryn, L.; et al. O03: Hydroxocobalamin (OHCbl) dose intensification can prevent visual deterioration and improve neurological and biochemical outcomes in CBLC deficiency. Genet. Med. Open 2025, 3 (Suppl. 2), 101966. [Google Scholar] [CrossRef]
- Bourque, D.K.; Mellin-Sanchez, L.E.; Bullivant, G.; Cruz, V.; Feigenbaum, A.; Hewson, S.; Raiman, J.; Schulze, A.; Siriwardena, K.; Mercimek-Andrews, S. Outcomes of patients with cobalamin C deficiency: A single center experience. JIMD Rep. 2020, 57, 102–114. [Google Scholar] [CrossRef]

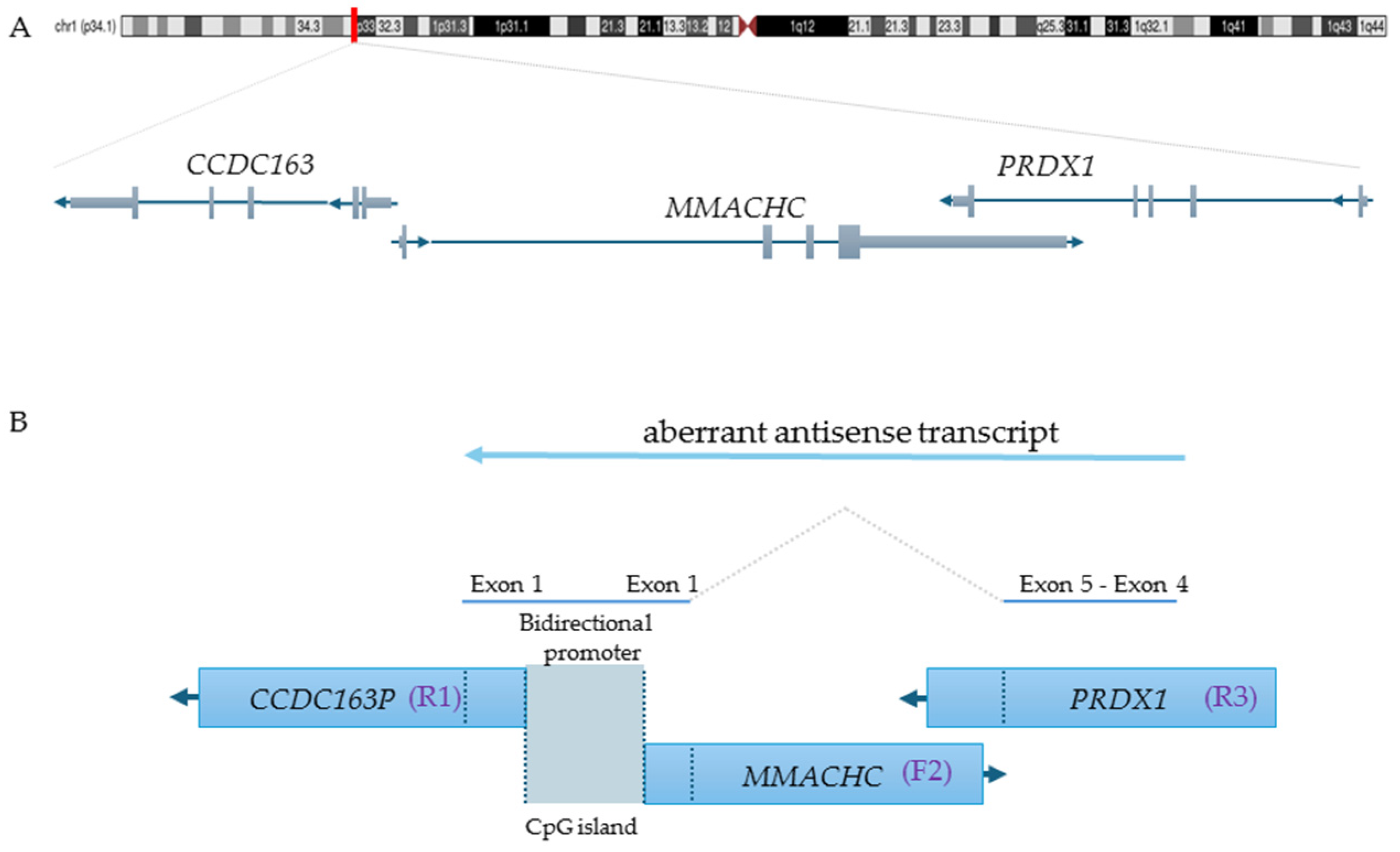

{kind=link}
{kind=link}
| Time | C3 (µm/L) | MMA (µm/L) | Hcy (µm/L) | Met (µm/L) | Therapy |
|---|---|---|---|---|---|
| 2 d (1st DBS) | 10.05 (nv < 3.3) | 66 (nv < 3.0) | 51.15 (nv < 10) | - | / |
| 5 d (2nd DBS) | 08.41 (nv < 3.3) | 101.6 (nv <3.0) | 57 (nv < 10) | 4 (nv 9–40) | / |
| 8 d | 7.76 (nv < 2.5) | 75.6 (nv < 5) | 230 (nv 5–15) | 9 (nv 9–39) | / |
| 9 d | - | - | 240 (nv 5–15) | - | OHCbl im 1 mg, betaine 100 mg × 3 daily, folic acid 5 mg × 2 weekly |
| 12 d | 0.83 (nv < 2.5) | 14.5 (nv < 5) | 72 (nv 5–15) | 27 (nv 9–39) | same as above |
| 20 d | 0.81 (nv < 2.5) | 4.7 (nv < 5) | 36 (nv 5–15) | 25 (nv 9–39) | same as above |
| 1 m 5 d | 1.19 (nv < 2.5) | 3.6 (nv < 5) | 36 (nv 5–15) | 47 (nv 9–39) | OHCbl (0.7 mL solution daily) with i-port advanceTM, betaine 300 mg × 3 daily, folic acid 5 mg × 2 weekly |
| 1 m 28 d | 0.81 (nv < 2.5) | 2.3 (nv < 5) | - | 30 (nv 9–39) | same as above |
| 3 m 1 d | - | - | 28 (nv 5–15) | - | same as above |
| 3 m 20 d | - | - | 18 (nv 5–15) | - | same as above |
| 5 m 9 d | 0.46 (nv < 2.5) | 1.5 (vn < 5) | 15.7 (nv < 12) | 19 (nv 9–39) | same as above |
| 7m 11 d | - | - | 21 (nv 5–15) | - | OHCbl im 5 mg (1 mL solution daily), betaine 400 mg × 3 daily, folic acid 5 mg × 2 weekly |
| 10 m 1 d | - | - | 24 (nv 5–15) | 33 (nv 9–42) | OHCbl im 5.5 mg (1.5 mL solution daily), betaine 500 mg × 3 daily, folic acid 5 mg × 2 weekly |
| a. Subgroup of Patients with Maculopathy Without Nystagmus | ||||||||||
| Age at First Retinal Exam | Retinal Findings (Fundus) and Age | OCT | ERG | Nyst | Strab | Optic Atrophy | Treatment Start Day | Visual Outcomes At Last Follow-Up | Study Design | Reference |
| 9 d | Mild pigmentary perifoveal dystrophy (3 m); central macular atrophy, slightly pale optic disc (5 m); BEM (7 m) | Foveal thinning at OPL (7 m) | Reduction of scotopic and photopic components (7 m) | - | - | - | 8 | FF | Prospective | This study, F |
| 4 m | BEM with progressive macular atrophy progressing between 4–7 m; peripheral non-perfusion | Progressive OPL and ONL atrophy to virtual disappearance of these layers in the fovea between 4–7 m | Abnormal | - | - | - | 4 | FF (8 m) | Retrospective and prospective | [4] (P56, M) |
| 6 w | BEM at 6 w | NR | NR | - | - | - | 8 | FF (6 w) | Retrospective and prospective | [4] (P57, M) |
| 6 m | Maculopathy | NR | Grossly normal at 12 m; diminished photopic and absent scotopic responses at 3 y; complete flattening at 10 y | - | NR | + | IAD | 3/200 (10 y) | Retrospective | [8] (P8, M; P53 in [4]) |
| 6 m | Maculopathy (BEM at 6 m; bone spicules; progression of the macular lesions, new mid- and far-peripheral RPE mottling in a geographic fashion at 2.8 y; TDP and bone spicules at 9.8 y) | Staphyloma in the central area of the chorioretinal atrophy (9.8 y) | Significant decrease in scotopic responses, moderate decrease in photopic responses (9.8 y) | - | - | + (9.8 y) | <1 m | 20/150 (7.3 y) | Retrospective and prospective | [11] (P4, F) |
| b. Subgroup of patients with maculopathy and nystagmus | ||||||||||
| Age at first retinal exam | Retinal findings (fundus) and age | OCT | ERG | Nyst | Strab | Optic atrophy | Treatment start day | Visual outcomes at last follow-up | Study design | Reference |
| 1 m | Maculopathy | NR | Abnormal photopic and scotopic | + | NR | - | IAD | 20/80 (10 y) | Retrospective | [8] (P2, M; P47 in [4]) |
| 7 m | Maculopathy (Faint BEM at 7 m, stable to 2 y) | NR | NR | + | - | - | Prenatal | CSM | Retrospective and prospective | [11] (P3, M) |
| NR | Maculopathy at 3 y | NR | NR | + | - | - | Prenatal | ‘low vision’ (14 m) | Retrospective and prospective | [4] (P59, M) |
| NR | Maculopathy at 3 y | NR | NR | + | - | - | <1 m | 20/100–20/50 | Retrospective | [22] (M; P32 in [4]) |
| NR | Maculopathy; RPE lucency and pigmentary retinopathy at 4 y | NR | NR | + | - | + | <1 m | CSM | Retrospective | [23] (P4, M; P36 in [4]) |
| NR | Maculopathy; bilateral central retinal thinning with gray pigmentation at 4 y | NR | NR | + | + | - | <1 m | FF | Retrospective | [23] (P5, M; P37 in [4]) |
| NR | Maculopathy with atrophy at 3 y; myopia | NR | NR | + | - | NR (poor VEP responses to flash and pattern stimulation) | <1 m | LA | Retrospective | [23] (P8, M; P40 in [4]) |
| c. Subgroup of patients with nystagmus without maculopathy | ||||||||||
| Age at first retinal exam | Retinal findings (fundus) and age | OCT | ERG | Nyst | Strab | Optic atrophy | Treatment start day | Visual outcomes at last follow-up | Study design | Reference |
| 5 m | Unremarkable fundus | NR | NR | + | - | - | <1 m | CSM | Retrospective and prospective | [11] (P5, M) |
| 8 m | BEM from 4.8 y, stable at last follow-up (7.3 y) | Parafoveal atrophy of outer retinal structures, consistent with BEM (4.8 y); worsening foveal and parafoveal atrophy of the outer retina and RPE, along with subretinal debris | Mildly decreased amplitude for rod and cone function (4.8 y, and stable at 6.8 y) | + (from 8 m) | - | + (5.4 y) | <1 m | 20/125 (7.3 y) | Retrospective and prospective | [11] (P6) |
| 6 m | Normal at 6 m | NR | Decreased photopically, extinguished scotopically | + (intermittent, horizontal and pendular, with occasional vertical component) | - | - | <1 m | CSM (6 m) | Prospective | [24] (P3, F; P24 in [4]) |
| 24 m | Normal at 24 m. Absent maculopathy; peripheral ‘salt and pepper’ retinopathy, normal discs at 14 y | NR | Scotopic normal, photopic slightly abnormal (decreased b-wave amplitude) at 14 y | + (Kestenbaum surgery at 5 y) | - | - | ? | 20/40 (14 y) | Retrospective | [25] (P3, M; P31 in [4]) |
| NR | Absent maculopathy; mottling of RPE with small dark central clump of pigmentation (7 y) | NR | Normal | + | - | + | <1 m | FF (7 y) | Retrospective | [23] (P1, M; P33 [4]) |
| NR | Normal at 3 y | NR | Normal | + | - | - | <1 m | FF (3 y) | Retrospective | [23] (P9, M; P41, in [4]) |
| 5 m | Normal at 11 y | NR | Normal | + | NR | + | IAD | 20/100 (11 y) | Retrospective | [8] (P5, F; P50 in [4]) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michieletto, P.; Baldo, F.; Madonia, M.; Zupin, L.; Pensiero, S.; Bonati, M.T. Retinal Changes in Early-Onset cblC Methylmalonic Acidemia Identified Through Expanded Newborn Screening: Highlights from a Case Study and Literature Review. Genes 2025, 16, 635. https://doi.org/10.3390/genes16060635
Michieletto P, Baldo F, Madonia M, Zupin L, Pensiero S, Bonati MT. Retinal Changes in Early-Onset cblC Methylmalonic Acidemia Identified Through Expanded Newborn Screening: Highlights from a Case Study and Literature Review. Genes. 2025; 16(6):635. https://doi.org/10.3390/genes16060635
Chicago/Turabian StyleMichieletto, Paola, Francesco Baldo, Maurizio Madonia, Luisa Zupin, Stefano Pensiero, and Maria Teresa Bonati. 2025. "Retinal Changes in Early-Onset cblC Methylmalonic Acidemia Identified Through Expanded Newborn Screening: Highlights from a Case Study and Literature Review" Genes 16, no. 6: 635. https://doi.org/10.3390/genes16060635
APA StyleMichieletto, P., Baldo, F., Madonia, M., Zupin, L., Pensiero, S., & Bonati, M. T. (2025). Retinal Changes in Early-Onset cblC Methylmalonic Acidemia Identified Through Expanded Newborn Screening: Highlights from a Case Study and Literature Review. Genes, 16(6), 635. https://doi.org/10.3390/genes16060635