
Colorectal Cancer Biomarker Identification via Joint DNA-Methylation and Transcriptomics Analysis Workflow


Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Identifying and Mapping Differentially Methylated CpG Sites
2.3. Normalization and Filtering
2.4. Identification of Differentially Expressed Genes (DEGs)
2.5. Statistical Methods for Differential Analyzes
2.6. Identification and Analysis of Methylation-Regulated Genes (MRGs)
2.7. Validation and Functional Enrichment of Methylation-Regulated Genes
3. Results
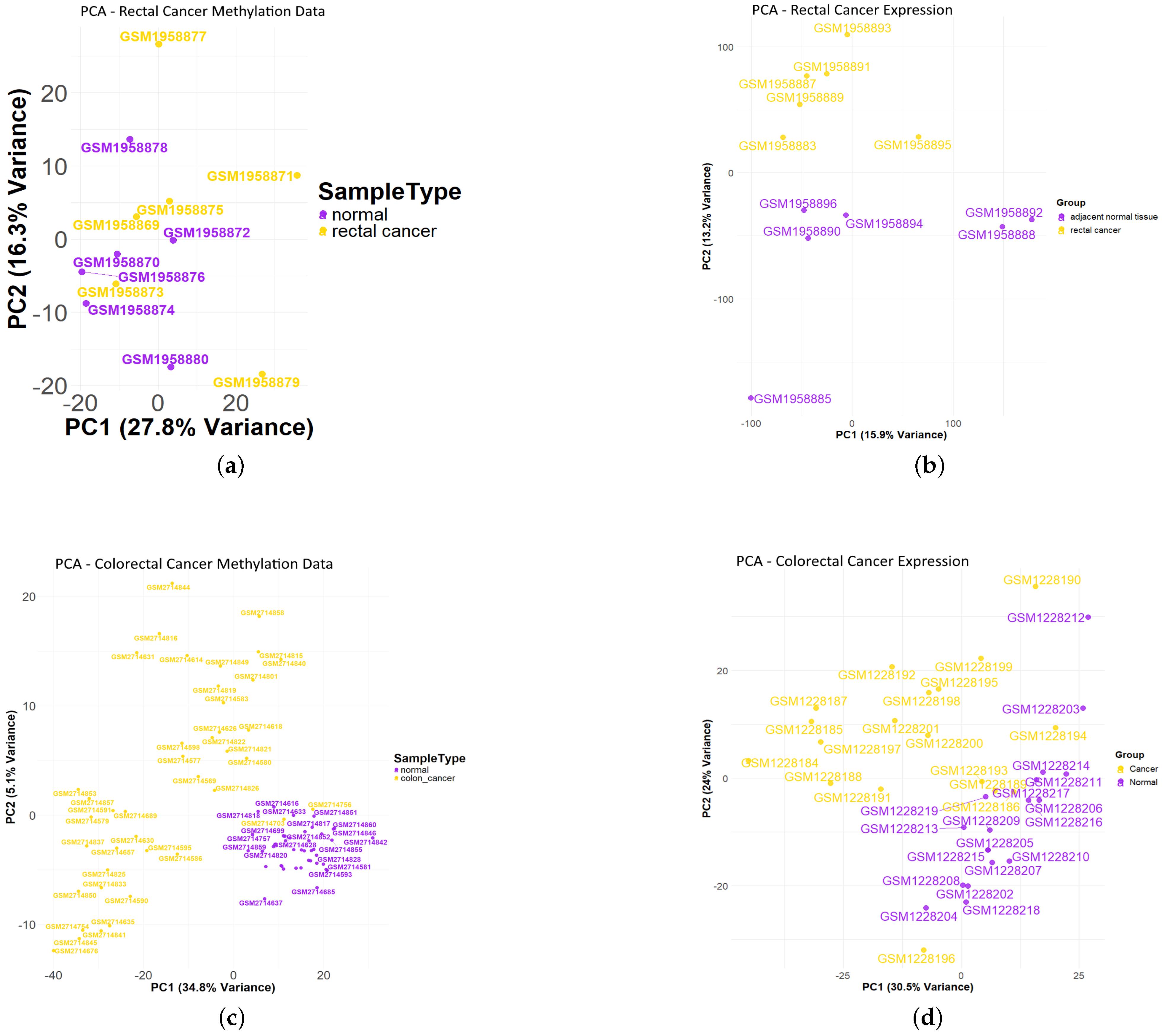
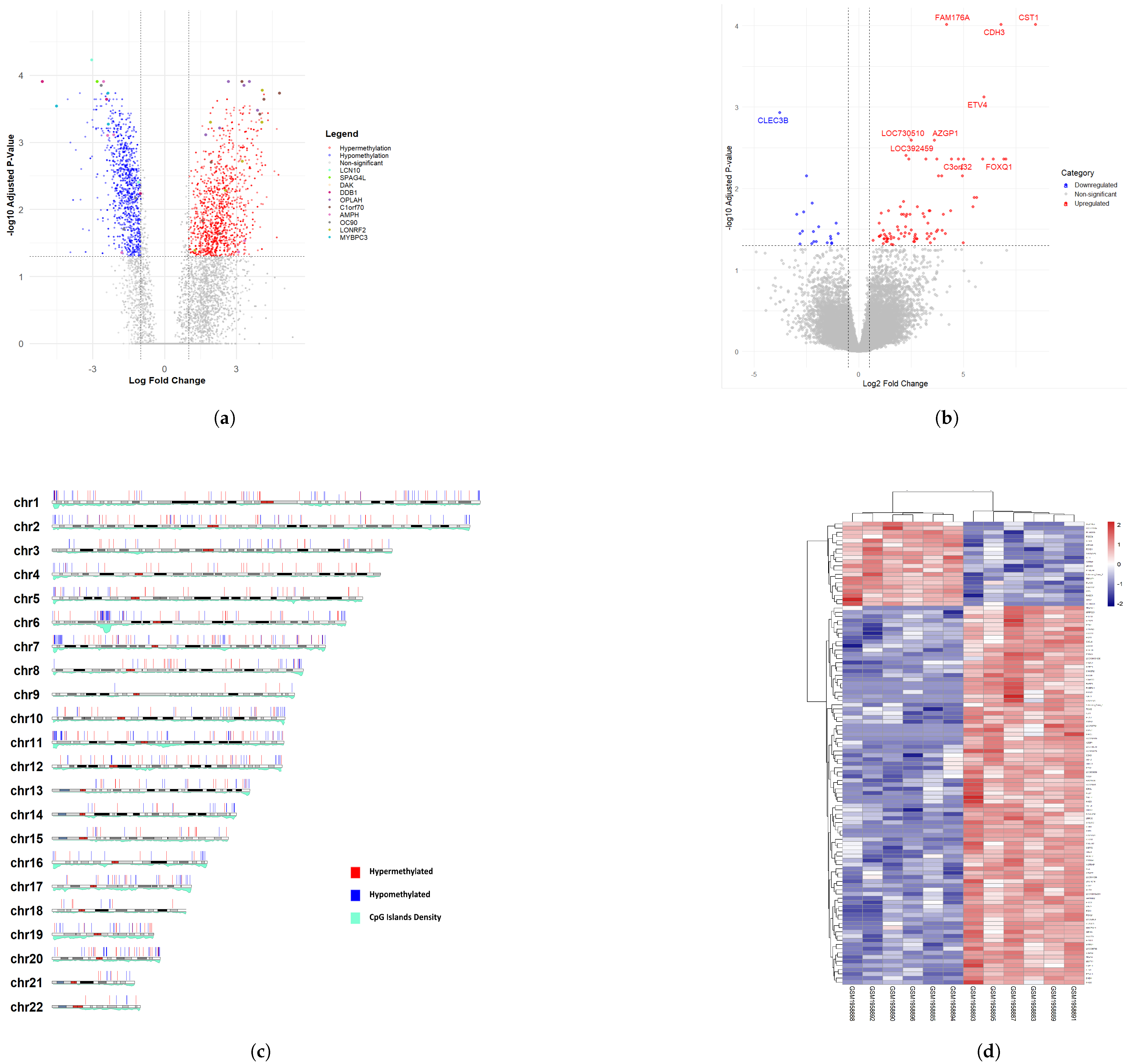
3.1. Differentially Methylated CpG Sites (DMCs) and Differentially Expressed Genes (DEGs) of Rectal Cancer Cohort
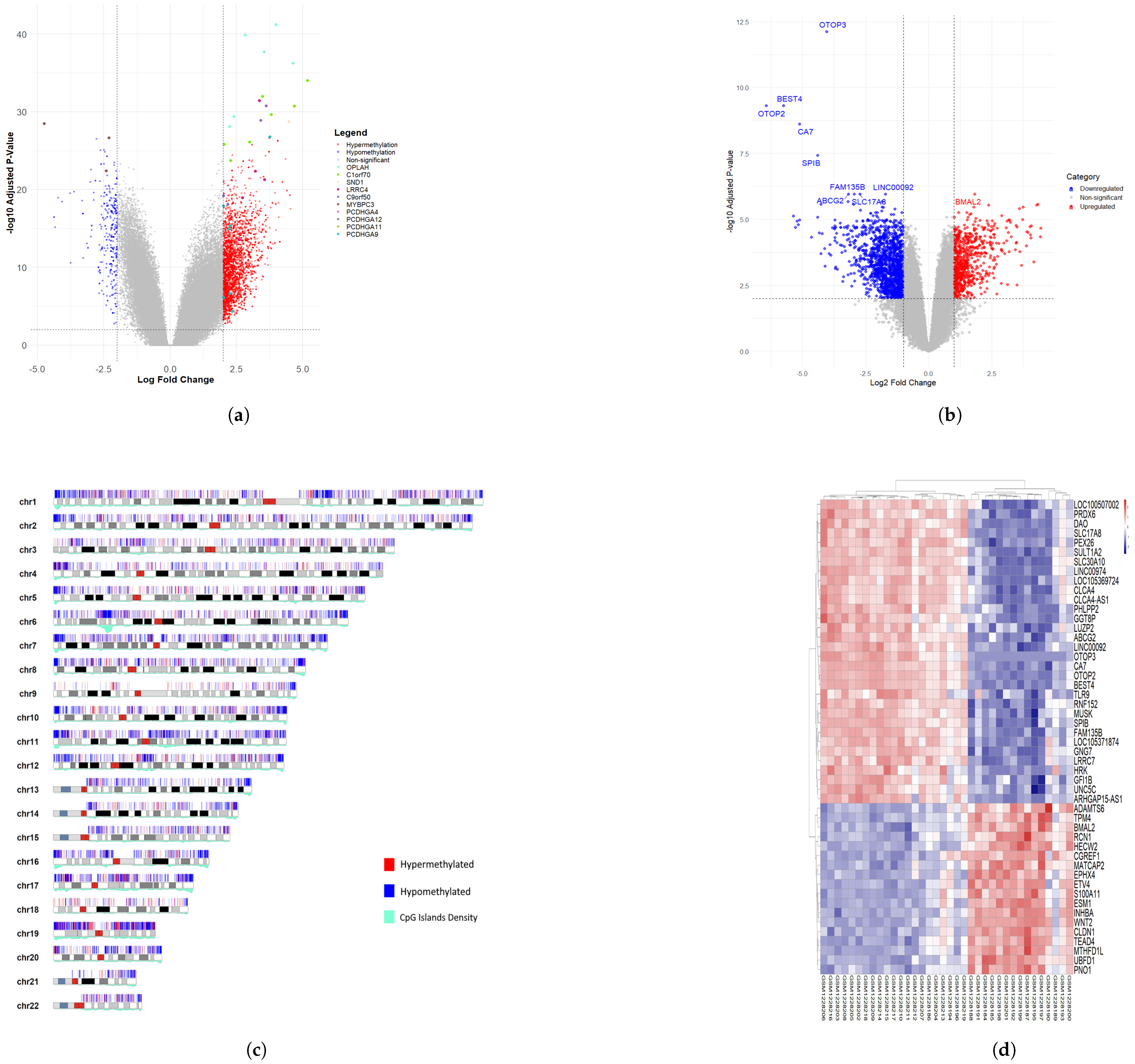
3.2. Differentially Methylated CpG Sites (DMCs) and Differentially Expressed Genes (DEGs) of Colon Cancer Cohort
3.3. Methylation-Regulated Genes (MRGs)
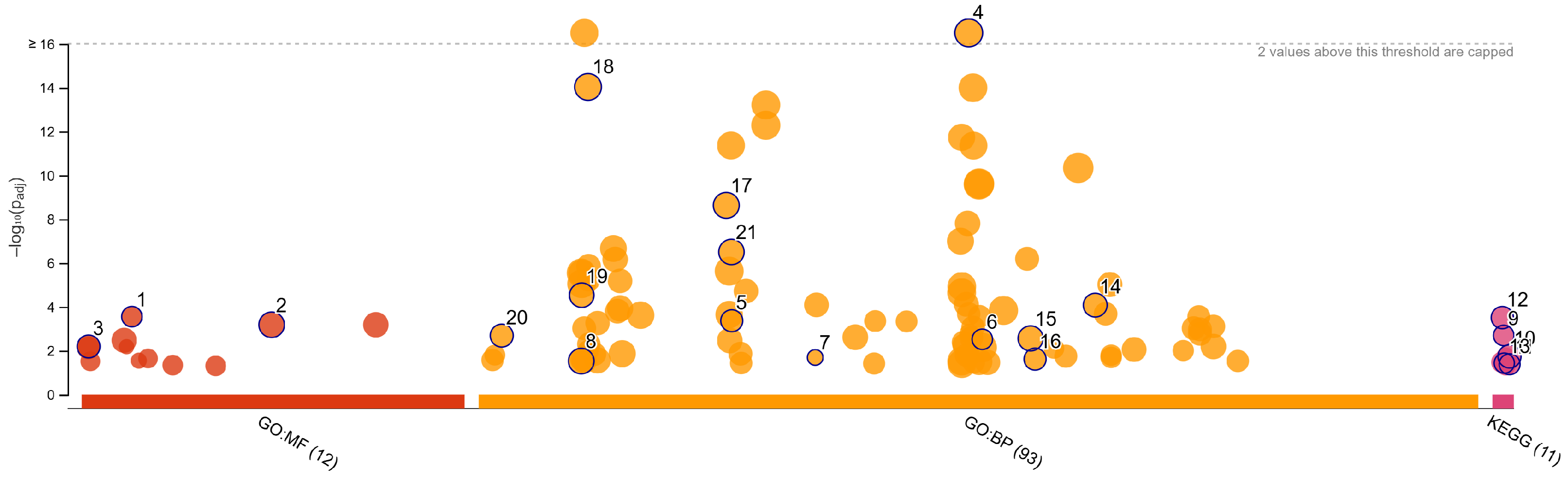
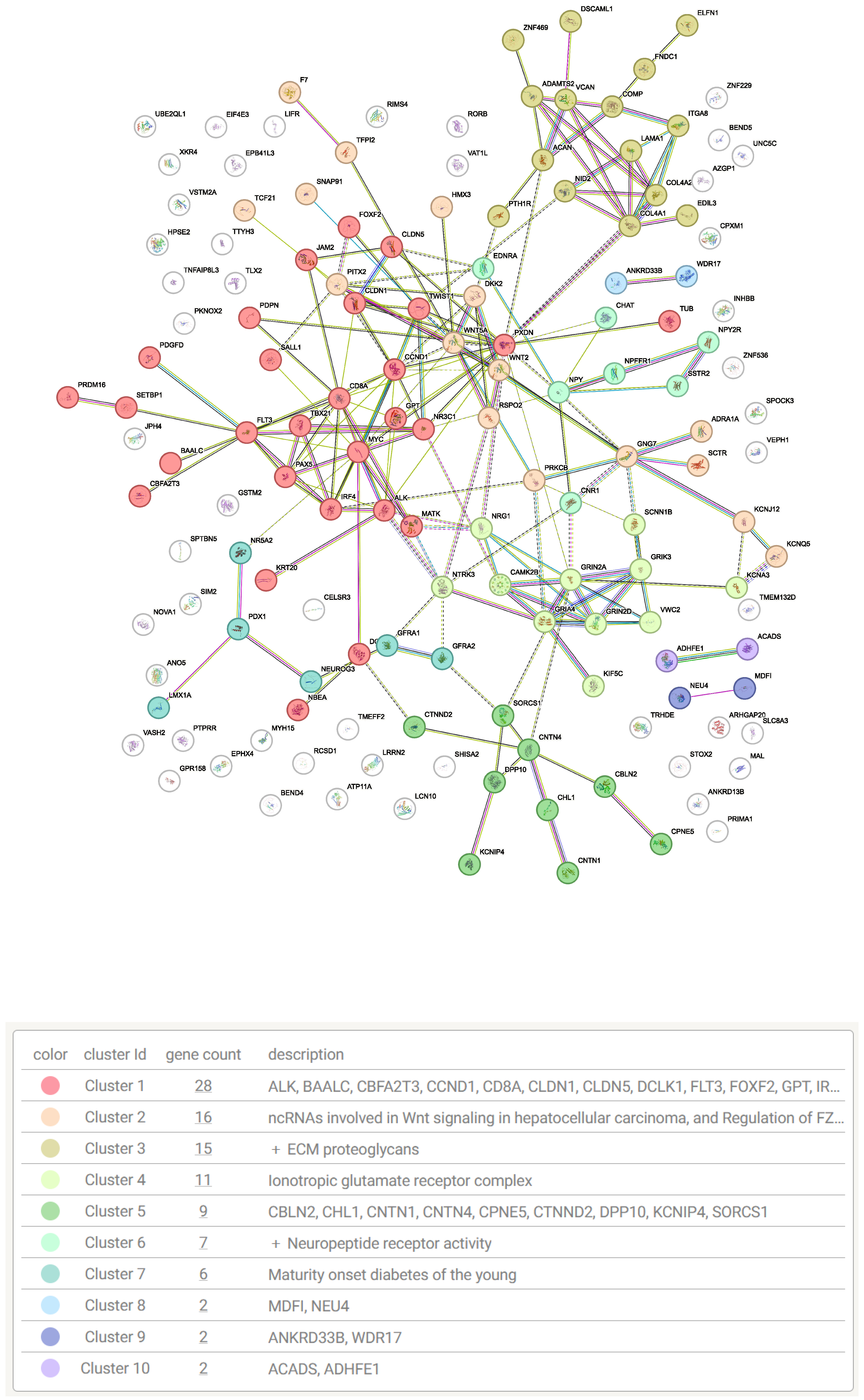
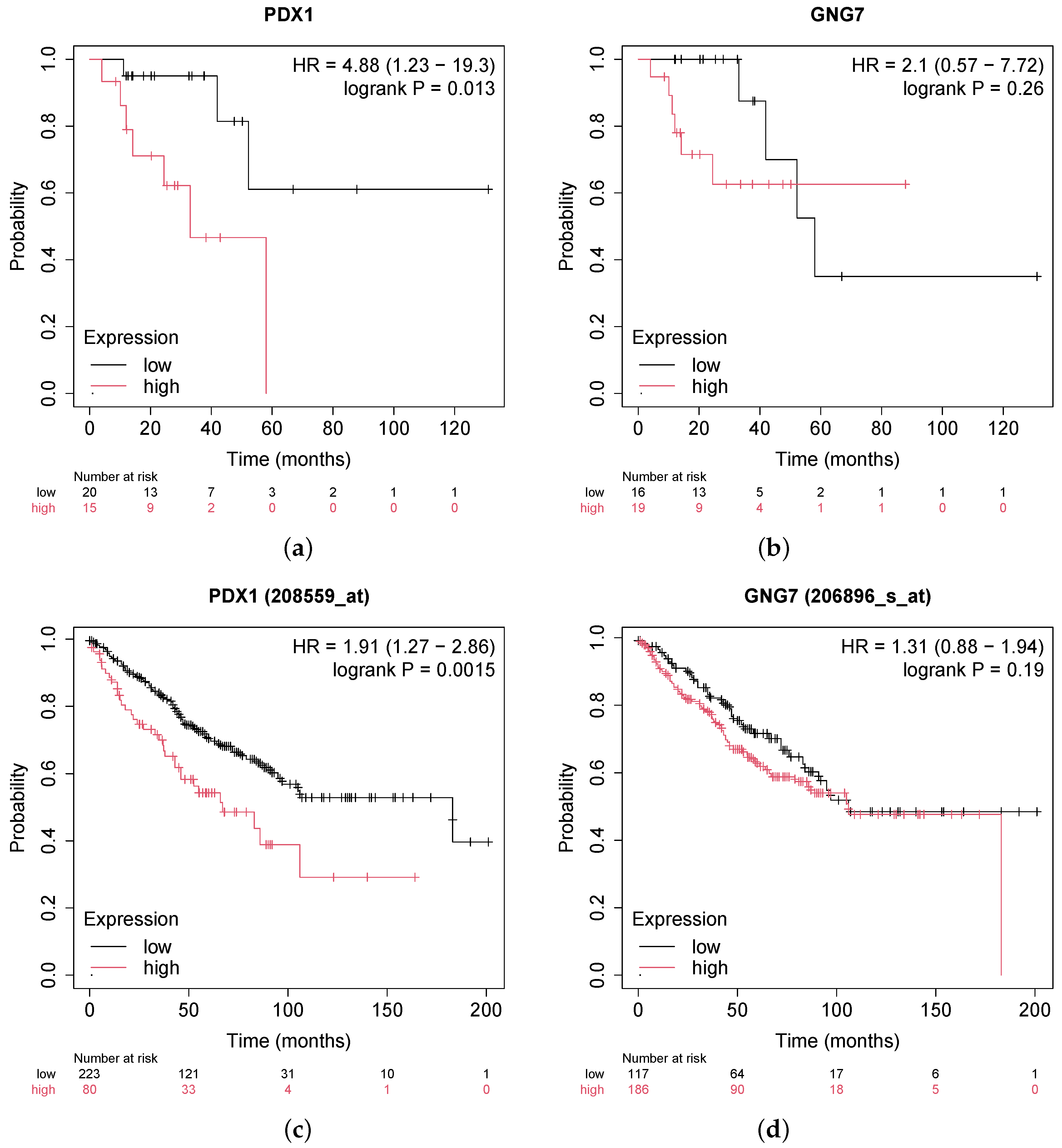
3.4. Validation and Functional Enrichment of Methylation-Regulated Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| CRC | Colorectal cancer |
| MRGs | Methylation-regulated genes |
| DMCs | Differentially methylated CpG sites |
| DMRs | Differentially methylated regions |
| DEGs | Differentially expressed genes |
References
- Alzahrani, S.M.; Al Doghaither, H.A.; Al-Ghafari, A.B. General insight into cancer: An overview of colorectal cancer. Mol. Clin. Oncol. 2021, 15, 271. [Google Scholar] [CrossRef] [PubMed]
- Hoang, T.; Kim, H.; Kim, J. Dietary intake in association with all-cause mortality and colorectal cancer mortality among colorectal cancer survivors: A systematic review and meta-analysis of prospective studies. Cancers 2020, 12, 3391. [Google Scholar] [CrossRef] [PubMed]
- Housini, M.; Dariya, B.; Ahmed, N.; Stevens, A.; Fiadjoe, H.; Nagaraju, G.P.; Basha, R. Colorectal cancer: Genetic alterations, novel biomarkers, current therapeutic strategies and clinical trials. Gene 2024, 892, 147857. [Google Scholar] [CrossRef]
- Ogunwobi, O.O.; Mahmood, F.; Akingboye, A. Biomarkers in colorectal cancer: Current research and future prospects. Int. J. Mol. Sci. 2020, 21, 5311. [Google Scholar] [CrossRef] [PubMed]
- Zygulska, A.L.; Pierzchalski, P. Novel diagnostic biomarkers in colorectal cancer. Int. J. Mol. Sci. 2022, 23, 852. [Google Scholar] [CrossRef]
- Vrba, L.; Futscher, B.W. DNA methylation changes in biomarker loci occur early in cancer progression. F1000Research 2020, 8, 2106. [Google Scholar] [CrossRef]
- Shen, Y.; Wang, D.; Yuan, T.; Fang, H.; Zhu, C.; Qin, J.; Xu, X.; Zhang, C.; Liu, J.; Zhang, Y.; et al. Novel DNA methylation biomarkers in stool and blood for early detection of colorectal cancer and precancerous lesions. Clin. Epigenet. 2023, 15, 26. [Google Scholar] [CrossRef]
- Zhao, G.; Liu, X.; Liu, Y.; Li, H.; Ma, Y.; Li, S.; Zhu, Y.; Miao, J.; Xiong, S.; Fei, S.; et al. Aberrant DNA methylation of SEPT9 and SDC2 in stool specimens as an integrated biomarker for colorectal cancer early detection. Front. Genet. 2020, 11, 643. [Google Scholar] [CrossRef]
- Nikolouzakis, T.K.; Vassilopoulou, L.; Fragkiadaki, P.; Mariolis Sapsakos, T.; Papadakis, G.Z.; Spandidos, D.A.; Tsatsakis, A.M.; Tsiaoussis, J. Improving diagnosis, prognosis and prediction by using biomarkers in CRC patients. Oncol. Rep. 2018, 39, 2455–2472. [Google Scholar] [CrossRef]
- Li, S.; Liu, Y.; Liu, M.; Wang, L.; Li, X. Comprehensive bioinformatics analysis reveals biomarkers of DNA methylation-related genes in varicose veins. Front. Genet. 2022, 13, 1013803. [Google Scholar] [CrossRef]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Peters, T.J.; Buckley, M.J.; Statham, A.L.; Pidsley, R.; Samaras, K.; V Lord, R.; Clark, S.J.; Molloy, P.L. De novo identification of differentially methylated regions in the human genome. Epigenet. Chromatin 2015, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Team TBD. BSgenome.Hsapiens.UCSC.hg19: Full Genome Sequences for Homo Sapiens (UCSC Version hg19, Based on GRCh37.p13), R package version 1.4.3; Team TBD: Houston, TX, USA, 2020. [Google Scholar]
- Gel, B.; Serra, E. karyoploteR: An R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 2017, 33, 3088–3090. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g: Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Lánczky, A.; Györffy, B. Web-based survival analysis tool tailored for medical research (KMplot): Development and implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef]
- Lewandowska, A.; Rudzki, G.; Lewandowski, T.; Stryjkowska-Gora, A.; Rudzki, S. Risk factors for the diagnosis of colorectal cancer. Cancer Control 2022, 29, 10732748211056692. [Google Scholar] [CrossRef]
- Raut, J.R.; Guan, Z.; Schrotz-King, P.; Brenner, H. Fecal DNA methylation markers for detecting stages of colorectal cancer and its precursors: A systematic review. Clin. Epigenet. 2020, 12, 122. [Google Scholar] [CrossRef]
- Bach, S.; Paulis, I.; Sluiter, N.; Tibbesma, M.; Martin, I.; Van De Wiel, M.; Tuynman, J.; Bahce, I.; Kazemier, G.; Steenbergen, R. Detection of colorectal cancer in urine using DNA methylation analysis. Sci. Rep. 2021, 11, 2363. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Li, Q.; Tu, X.; Yu, D.; Zhou, Y.; Ma, L.; Wei, K.; Gao, Y.; Zhao, G.; Han, R.; et al. DNA hypomethylation patterns and their impact on the tumor microenvironment in colorectal cancer. Cell. Oncol. 2024, 47, 1375–1389. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Yin, R.X.; Zhang, Q.H.; Hu, X.J.; Huang, F.; Chen, W.X.; Cao, X.L.; Wu, J.Z. Integrated DNA methylation and gene expression analysis in the pathogenesis of coronary artery disease. Aging 2019, 11, 1486. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yi, J.; Yang, J.; Han, Y.; Qian, X.; Liu, Y.; Li, J.; Lu, B.; Zhang, J.; Pan, X.; et al. An integrated epigenomic-transcriptomic landscape of lung cancer reveals novel methylation driver genes of diagnostic and therapeutic relevance. Theranostics 2021, 11, 5346. [Google Scholar] [CrossRef]
- Liu, J.; Li, H.; Sun, L.; Wang, Z.; Xing, C.; Yuan, Y. Aberrantly methylated-differentially expressed genes and pathways in colorectal cancer. Cancer Cell Int. 2017, 17, 75. [Google Scholar] [CrossRef]
- Sun, G.; Li, Y.; Peng, Y.; Lu, D.; Zhang, F.; Cui, X.; Zhang, Q.; Li, Z. Identification of differentially expressed genes and biological characteristics of colorectal cancer by integrated bioinformatics analysis. J. Cell. Physiol. 2019, 234, 15215–15224. [Google Scholar] [CrossRef]
- Wei, Q.; Miao, T.; Zhang, P.; Jiang, B.; Yan, H. Comprehensive analysis to identify GNG7 as a prognostic biomarker in lung adenocarcinoma correlating with immune infiltrates. Front. Genet. 2022, 13, 984575. [Google Scholar] [CrossRef]
- Ebrahim, N.; Shakirova, K.; Dashinimaev, E. PDX1 is the cornerstone of pancreatic β-cell functions and identity. Front. Mol. Biosci. 2022, 9, 1091757. [Google Scholar] [CrossRef]
- Vinogradova, T.; Sverdlov, E. PDX1: A unique pancreatic master regulator constantly changes its functions during embryonic development and progression of pancreatic cancer. Biochemistry 2017, 82, 887–893. [Google Scholar] [CrossRef]
- Lee, Y.; Dho, S.H.; Lee, J.; Hwang, J.H.; Kim, M.; Choi, W.Y.; Lee, J.Y.; Lee, J.; Chang, W.; Lee, M.Y.; et al. Hypermethylation of PDX1, EN2, and MSX1 predicts the prognosis of colorectal cancer. Exp. Mol. Med. 2022, 54, 156–168. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, X. Advances of Wnt signalling pathway in colorectal cancer. Cells 2023, 12, 447. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Geng, S.; Luo, H.; Wang, W.; Mo, Y.Q.; Luo, Q.; Wang, L.; Song, G.B.; Sheng, J.P.; Xu, B. Signaling pathways involved in colorectal cancer: Pathogenesis and targeted therapy. Signal Transduct. Target. Ther. 2024, 9, 266. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, S.; Nyström, H. The extracellular matrix in colorectal cancer and its metastatic settling–Alterations and biological implications. Crit. Rev. Oncol. /Hematol. 2022, 175, 103712. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Ha, S.E.; Wu, M.; Zogg, H.; Ronkon, C.F.; Lee, M.Y.; Ro, S. Extracellular matrix biomarkers in colorectal cancer. Int. J. Mol. Sci. 2021, 22, 9185. [Google Scholar] [CrossRef]
- Holland, A.M.; Bon-Frauches, A.C.; Keszthelyi, D.; Melotte, V.; Boesmans, W. The enteric nervous system in gastrointestinal disease etiology. Cell. Mol. Life Sci. 2021, 78, 4713–4733. [Google Scholar] [CrossRef]
- Battaglin, F.; Jayachandran, P.; Strelez, C.; Lenz, A.; Algaze, S.; Soni, S.; Lo, J.H.; Yang, Y.; Millstein, J.; Zhang, W.; et al. Neurotransmitter signaling: A new frontier in colorectal cancer biology and treatment. Oncogene 2022, 41, 4769–4778. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, L.; Jiang, S.; Yu, M. Nerve dependence in colorectal cancer. Front. Cell Dev. Biol. 2022, 10, 766653. [Google Scholar] [CrossRef]
- Liu, J.; Lang, G.; Shi, J. Epigenetic regulation of PDX-1 in type 2 diabetes mellitus. Diabetes Metab. Syndr. Obes. 2021, 14, 431–442. [Google Scholar] [CrossRef]
- Cheng, H.C.; Chang, T.K.; Su, W.C.; Tsai, H.L.; Wang, J.Y. Narrative review of the influence of diabetes mellitus and hyperglycemia on colorectal cancer risk and oncological outcomes. Transl. Oncol. 2021, 14, 101089. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RC MRGs | ||||||||
|---|---|---|---|---|---|---|---|---|
| Gene | logFC (Expr) | AveExpr (Expr) | t (Expr) | p.Value (Expr) | Adj.P.Val (Expr) | B (Expr) | Up- or Downregulation | Hyper- or Hypomethylation |
| GNG7 | −2.02633 | 4.84152 | −5.95184 | 0.04456 | 1.72179 | Down | Hypo | |
| HKDC1 | 3.60053 | 5.90876 | 6.04456 | 0.04150 | 1.84160 | Up | Hypo | |
| AZGP1 | 3.60273 | 2.10376 | 10.7325 | 0.00255 | 6.29499 | Up | Hypo | |
| ALG1L | 3.05863 | 1.81574 | 7.23563 | 0.02028 | 3.25665 | Up | Hypo | |
| PITX2 | 3.70735 | 2.94234 | 6.50273 | 0.03376 | 2.41281 | Up | Hyper | |
| PDX1 | 4.37360 | 3.77735 | 7.36968 | 0.01871 | 3.40209 | Up | Hyper | |
| CRC MRGs | ||||||||
| WNT2 | 4.33366 | −0.08931 | 7.87975 | 11.45417 | Up | Hyper | ||
| UNC5C | −1.79796 | 2.91492 | −7.67632 | 10.87752 | Down | Hyper | ||
| CLDN1 | 3.60033 | 2.97942 | 7.62796 | 10.73974 | Up | Hypo | ||
| GNG7 | −2.20229 | 2.97970 | −7.24621 | 9.64326 | Down | Hypo | ||
| EPHX4 | 2.89609 | −0.17779 | 7.23503 | 9.61091 | Up | Hypo | ||
| PDPN | 2.03958 | 3.26737 | 6.94714 | 8.77430 | Up | Hyper | ||
| TRHDE | −1.74839 | 1.34963 | −6.77779 | 8.27887 | Down | Hyper | ||
| CPNE5 | −1.31592 | 3.69191 | −6.64090 | 7.87689 | Down | Hyper | ||
| VWC2 | −1.58839 | −0.61997 | −6.47318 | 7.38269 | Down | Hyper | ||
| COL4A1 | 2.22504 | 7.58071 | 6.44237 | 7.29174 | Up | Hyper | ||
| Source | Term Name | Term ID | −log10 p | Intersect |
|---|---|---|---|---|
| GO:MF | extracellular matrix structural constituent | GO:0005201 | 3.5524 | 10 |
| GO:MF | signaling receptor activity | GO:0038023 | 3.1813 | 29 |
| GO:MF | DNA-binding transcription activator activity... | GO:0001228 | 2.1923 | 14 |
| GO:BP | system development | GO:0048731 | 16.3616 | 75 |
| GO:BP | nervous system development | GO:0007399 | 14.0332 | 57 |
| GO:BP | neurogenesis | GO:0022008 | 8.6293 | 40 |
| GO:BP | neuron differentiation | GO:0030182 | 6.5024 | 33 |
| GO:BP | enzyme-linked receptor protein signaling pathway | GO:0007167 | 4.5302 | 24 |
| GO:BP | response to growth factor | GO:0070848 | 4.0768 | 20 |
| GO:BP | extracellular matrix organization | GO:0030198 | 3.3660 | 13 |
| GO:BP | morphogenesis of an epithelium | GO:0002009 | 2.6869 | 15 |
| GO:BP | epithelium development | GO:0060429 | 2.5588 | 24 |
| GO:BP | neuromuscular process | GO:0050905 | 2.5201 | 9 |
| GO:BP | ionotropic glutamate receptor signaling pathway | GO:0035235 | 1.6912 | 4 |
| GO:BP | epithelial tube morphogenesis | GO:0060562 | 1.6144 | 11 |
| GO:BP | cell adhesion | GO:0007155 | 1.5264 | 25 |
| KEGG | Neuroactive ligand-receptor interaction | KEGG:04080 | 3.5077 | 14 |
| KEGG | Wnt signaling pathway | KEGG:04310 | 2.6935 | 9 |
| KEGG | Pathways in cancer | KEGG:05200 | 1.7382 | 14 |
| KEGG | Cell adhesion molecules | KEGG:04514 | 1.4320 | 7 |
| KEGG | Proteoglycans in cancer | KEGG:05205 | 1.3830 | 8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oladapo, O.B.; Moussa, M.R. Colorectal Cancer Biomarker Identification via Joint DNA-Methylation and Transcriptomics Analysis Workflow. Genes 2025, 16, 620. https://doi.org/10.3390/genes16060620
Oladapo OB, Moussa MR. Colorectal Cancer Biomarker Identification via Joint DNA-Methylation and Transcriptomics Analysis Workflow. Genes. 2025; 16(6):620. https://doi.org/10.3390/genes16060620
Chicago/Turabian StyleOladapo, Olajumoke B., and Marmar R. Moussa. 2025. "Colorectal Cancer Biomarker Identification via Joint DNA-Methylation and Transcriptomics Analysis Workflow" Genes 16, no. 6: 620. https://doi.org/10.3390/genes16060620
APA StyleOladapo, O. B., & Moussa, M. R. (2025). Colorectal Cancer Biomarker Identification via Joint DNA-Methylation and Transcriptomics Analysis Workflow. Genes, 16(6), 620. https://doi.org/10.3390/genes16060620