
The Role of Artificial Intelligence in Identifying NF1 Gene Variants and Improving Diagnosis
 ,
,
Abstract
1. Introduction
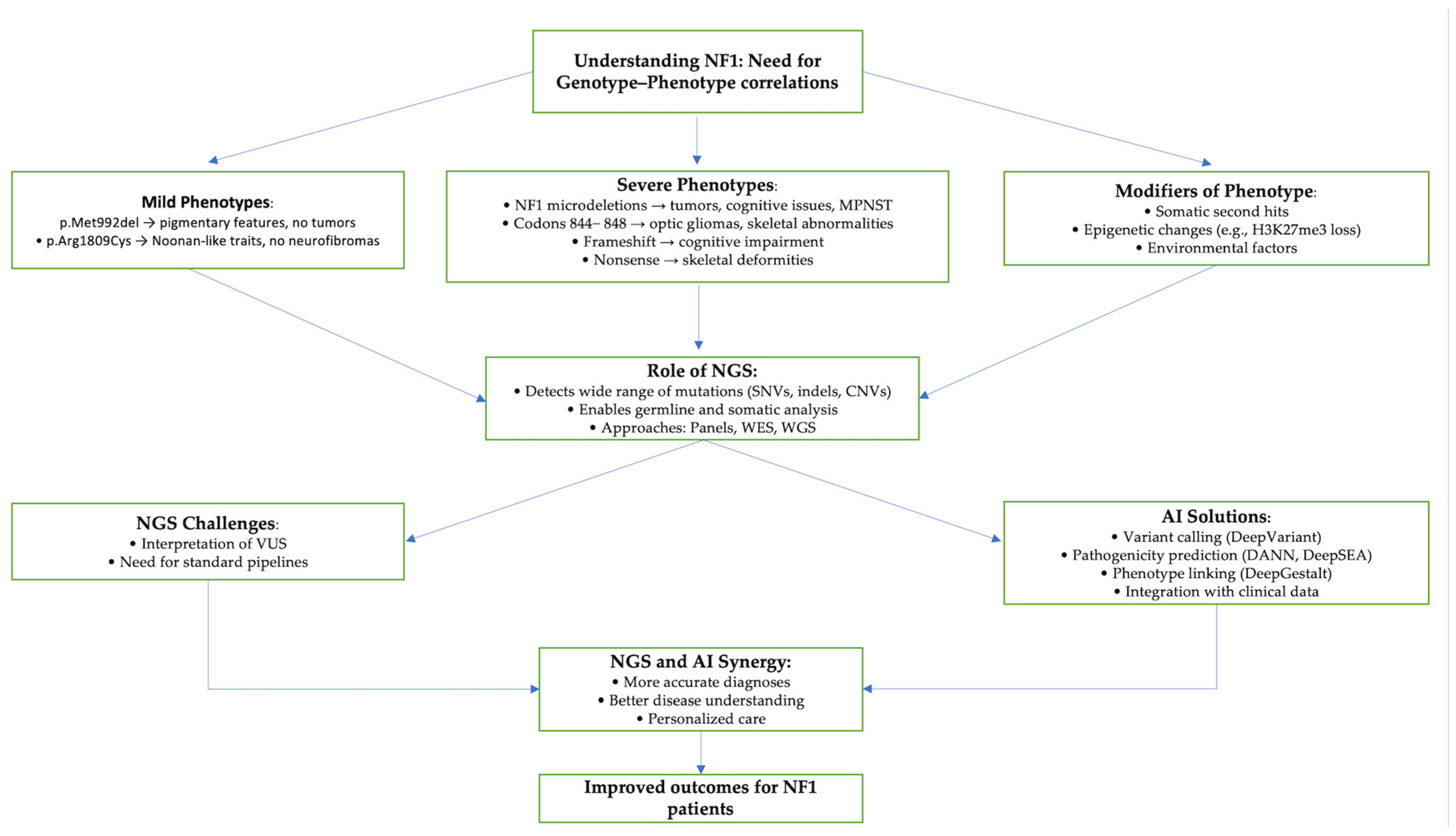
NF1: Genetic and Clinical Overview
2. Challenges in Genetic Testing and Variant Interpretation
3. AI in NF1 Variants
3.1. Variant Interpretation Tools
3.2. Predicting Pathogenicity of NF1 Variants
4. Clinical Pipelines
4.1. Tumor Detection
4.2. Therapeutic Prediction
5. Future Directions
5.1. NGS for Genotype–Phenotype Correlation
5.2. Multi-Omics
6. Bias of AI in Genetics
7. Strengths and Limitations
8. AI Diagnosing NF1 Beyond Genetics
9. Discussion
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Friedman, J.M. Neurofibromatosis 1. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2019; pp. 1993–2021. [Google Scholar]
- Wallace, M.R.; Marchuk, D.A.; Andersen, L.B.; Letcher, R.; Odeh, H.M.; Saulino, A.M.; Fountain, J.W.; Brereton, A.; Nicholson, J.; Mitchell, A.L.; et al. Type 1 neurofibromatosis gene: Identification of a large transcript disrupted in three NF1 patients. Science 1990, 249, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Arun, D.; Gutmann, D.H. Recent advances in neurofibromatosis type 1. Curr. Opin. Neurol. 2004, 17, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Longo, J.F.; Weber, S.M.; Turner-Ivey, B.P.; Carroll, S.L. Recent Advances in the Diagnosis and Pathogenesis of Neurofibromatosis Type 1 (NF1)-associated Peripheral Nervous System Neoplasms. Adv. Anat. Pathol. 2018, 25, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Uusitalo, E.; Leppävirta, J.; Koffert, A.; Suominen, S.; Vahtera, J.; Vahlberg, T.; Pöyhönen, M.; Peltonen, J.; Peltonen, S. Incidence and Mortality of Neurofibromatosis: A Total Population Study in Finland. J. Investig. Dermatol. 2015, 135, 904–906. [Google Scholar] [CrossRef]
- Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.A.; Berman, Y.; Blakeley, J.; Babovic-Vuksanovic, D.; Cunha, K.S.; Ferner, R.; et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet. Med. 2021, 23, 1506–1513. [Google Scholar] [CrossRef]
- Bergoug, M.; Doudeau, M.; Godin, F.; Mosrin, C.; Vallée, B.; Bénédetti, H. Neurofibromin Structure, Functions and Regulation. Cells 2020, 9, 2365. [Google Scholar] [CrossRef]
- Messiaen, L.; Xie, J. NF1 Germline and Somatic Mosaicism. In Neurofibromatosis Type 1: Molecular and Cellular Biology; Upadhyaya, M., Cooper, D.N., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 151–172. [Google Scholar]
- Pacot, L.; Vidaud, D.; Ye, M.; Chansavang, A.; Coustier, A.; Maillard, T.; Barbance, C.; Laurendeau, I.; Hebrard, B.; Lunati-Rozie, A.; et al. Prenatal diagnosis for neurofibromatosis type 1 and the pitfalls of germline mosaics. NPJ Genom. Med. 2024, 9, 41. [Google Scholar] [CrossRef]
- Pasmant, E.; Parfait, B.; Luscan, A.; Goussard, P.; Briand-Suleau, A.; Laurendeau, I.; Fouveaut, C.; Leroy, C.; Montadert, A.; Wolkenstein, P.; et al. Neurofibromatosis type 1 molecular diagnosis: What can NGS do for you when you have a large gene with loss of function mutations? Eur. J. Hum. Genet. 2015, 23, 596–601. [Google Scholar] [CrossRef]
- Tsipi, M.; Poulou, M.; Fylaktou, I.; Kosma, K.; Tsoutsou, E.; Pons, M.R.; Kokkinou, E.; Kitsiou-Tzeli, S.; Fryssira, H.; Tzetis, M. Phenotypic expression of a spectrum of Neurofibromatosis Type 1 (NF1) mutations identified through NGS and MLPA. J. Neurol. Sci. 2018, 395, 95–105. [Google Scholar] [CrossRef]
- Bildirici, Y.; Kocaaga, A.; Karademir-Arslan, C.N.; Yimenicioglu, S. Evaluation of Molecular and Clinical Findings in Children With Neurofibromatosis Type 1: Identification of 15 Novel Variants. Pediatr. Neurol. 2023, 149, 69–74. [Google Scholar] [CrossRef]
- Kim, S.H.; Kwon, S.S.; Park, M.R.; Lee, H.A.; Kim, J.H.; Cha, J.; Kim, S.; Baek, S.T.; Kim, S.H.; Lee, J.S.; et al. Detecting Low-Variant Allele Frequency Mosaic Pathogenic Variants of NF1, TSC2, and AKT3 Genes from Blood in Patients with Neurodevelopmental Disorders. J. Mol. Diagn. 2023, 25, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Koster, R.; Brandão, R.D.; Tserpelis, D.; van Roozendaal, C.E.P.; van Oosterhoud, C.N.; Claes, K.B.M.; Paulussen, A.D.C.; Sinnema, M.; Vreeburg, M.; van der Schoot, V.; et al. Pathogenic neurofibromatosis type 1 (NF1) RNA splicing resolved by targeted RNAseq. NPJ Genom. Med. 2021, 6, 95. [Google Scholar] [CrossRef]
- DeBella, K.; Szudek, J.; Friedman, J.M. Use of the national institutes of health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics 2000, 105, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Kehrer-Sawatzki, H.; Cooper, D.N. Classification of NF1 microdeletions and its importance for establishing genotype/phenotype correlations in patients with NF1 microdeletions. Hum. Genet. 2021, 140, 1635–1649. [Google Scholar] [CrossRef]
- Kehrer-Sawatzki, H.; Cooper, D.N. Challenges in the diagnosis of neurofibromatosis type 1 (NF1) in young children facilitated by means of revised diagnostic criteria including genetic testing for pathogenic NF1 gene variants. Hum. Genet. 2022, 141, 177–191. [Google Scholar] [CrossRef]
- Koczkowska, M.; Callens, T.; Chen, Y.; Gomes, A.; Hicks, A.D.; Sharp, A.; Johns, E.; Uhas, K.A.; Armstrong, L.; Bosanko, K.A.; et al. Clinical spectrum of individuals with pathogenic NF1 missense variants affecting p.Met1149, p.Arg1276, and p.Lys1423: Genotype-phenotype study in neurofibromatosis type 1. Hum. Mutat. 2020, 41, 299–315. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Frazer, J.; Notin, P.; Dias, M.; Gomez, A.; Min, J.K.; Brock, K.; Gal, Y.; Marks, D.S. Disease variant prediction with deep generative models of evolutionary data. Nature 2021, 599, 91–95. [Google Scholar] [CrossRef]
- de Sainte Agathe, J.M.; Filser, M.; Isidor, B.; Besnard, T.; Gueguen, P.; Perrin, A.; Van Goethem, C.; Verebi, C.; Masingue, M.; Rendu, J.; et al. SpliceAI-visual: A free online tool to improve SpliceAI splicing variant interpretation. Hum. Genom. 2023, 17, 7. [Google Scholar] [CrossRef]
- Ha, C.; Kim, J.W.; Jang, J.H. Performance Evaluation of SpliceAI for the Prediction of Splicing of NF1 Variants. Genes 2021, 12, 1308. [Google Scholar] [CrossRef]
- Huber, C.D.; Kim, B.Y.; Lohmueller, K.E. Population genetic models of GERP scores suggest pervasive turnover of constrained sites across mammalian evolution. PLoS Genet. 2020, 16, e1008827. [Google Scholar] [CrossRef]
- Lu, Q.; Hu, Y.; Sun, J.; Cheng, Y.; Cheung, K.H.; Zhao, H. A statistical framework to predict functional non-coding regions in the human genome through integrated analysis of annotation data. Sci. Rep. 2015, 5, 10576. [Google Scholar] [CrossRef]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2019, 48, D835–D844. [Google Scholar] [CrossRef]
- Liu, Y.; Yeung, W.S.B.; Chiu, P.C.N.; Cao, D. Computational approaches for predicting variant impact: An overview from resources, principles to applications. Front. Genet. 2022, 13, 981005. [Google Scholar] [CrossRef]
- Choon, Y.W.; Choon, Y.F.; Nasarudin, N.A.; Al Jasmi, F.; Remli, M.A.; Alkayali, M.H.; Mohamad, M.S. Artificial intelligence and database for NGS-based diagnosis in rare disease. Front. Genet. 2023, 14, 1258083. [Google Scholar] [CrossRef]
- Alirezaie, N.; Kernohan, K.D.; Hartley, T.; Majewski, J.; Hocking, T.D. ClinPred: Prediction Tool to Identify Disease-Relevant Nonsynonymous Single-Nucleotide Variants. Am. J. Hum. Genet. 2018, 103, 474–483. [Google Scholar] [CrossRef]
- Accetturo, M.; Bartolomeo, N.; Stella, A. In-silico Analysis of NF1 Missense Variants in ClinVar: Translating Variant Predictions into Variant Interpretation and Classification. Int. J. Mol. Sci. 2020, 21, 721. [Google Scholar] [CrossRef]
- Carter, H.; Douville, C.; Stenson, P.D.; Cooper, D.N.; Karchin, R. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genom. 2013, 14 (Suppl. 3), S3. [Google Scholar] [CrossRef]
- Chen, J.; Li, Z.; Wu, Y.; Li, X.; Chen, Z.; Chen, P.; Ding, Y.; Wu, C.; Hu, L. Identification of Pathogenic Missense Mutations of NF1 Using Computational Approaches. J. Mol. Neurosci. 2024, 74, 94. [Google Scholar] [CrossRef]
- Marino, S.M.; Gladyshev, V.N. Cysteine Function Governs Its Conservation and Degeneration and Restricts Its Utilization on Protein Surfaces. J. Mol. Biol. 2010, 404, 902–916. [Google Scholar] [CrossRef]
- Miller, D.T.; Lee, K.; Gordon, A.S.; Amendola, L.M.; Adelman, K.; Bale, S.J.; Chung, W.K.; Gollob, M.H.; Harrison, S.M.; Herman, G.E.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2021 update: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1391–1398. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Yang, E.-W.; Velazquez-Villarreal, E. AI-HOPE: An AI-Driven conversational agent for enhanced clinical and genomic data integration in precision medicine research. medRxiv 2025. [Google Scholar] [CrossRef]
- Kumar Mamidi, T.K.; Worthey, E.; Kaur, G.; Fay, C. O48: DITTO4NF: In silico classification and prioritization of likely pathogenic variants for NF1 using explainable machine learning. Genet. Med. Open 2023, 1, 100701. [Google Scholar] [CrossRef]
- Pagel, K.A.; Kim, R.; Moad, K.; Busby, B.; Zheng, L.; Hynes-Grace, M.; Tokheim, C.; Ryan, M.; Karchin, R. OpenCRAVAT, an open source collaborative platform for the annotation of human genetic variation. bioRxiv 2019, 794297. [Google Scholar] [CrossRef]
- Bidollahkhany, M.; Atasoy, F.; Abedini, E.; Davar, A.; Hamza, O.; Sefaoğlu, F.; Jafari, A.; Yalçın, M.; Abdellatef, H. GENIE-NF-AI: Identifying Neurofibromatosis Tumors using Liquid Neural Network (LTC) trained on AACR GENIE Datasets. arXiv 2023, arXiv:2304.13429. [Google Scholar]
- The AACR Project GENIE Consortium; André, F.; Arnedos, M.; Baras, A.S.; Baselga, J.; Bedard, P.L.; Berger, M.F.; Bierkens, M.; Calvo, F.; Cerami, E.; et al. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef]
- Bonetti, E.; Pellegatta, S.; Rosati, N.; Eoli, M.; Mazzarella, L. RENOVO-NF1 accurately predicts NF1 missense variant pathogenicity. medRxiv 2025. [Google Scholar] [CrossRef]
- Favalli, V.; Tini, G.; Bonetti, E.; Vozza, G.; Guida, A.; Gandini, S.; Pelicci, P.G.; Mazzarella, L. Machine learning-based reclassification of germline variants of unknown significance: The RENOVO algorithm. Am. J. Hum. Genet. 2021, 108, 682–695. [Google Scholar] [CrossRef]
- Martorana, D.; Barili, V.; Uliana, V.; Ambrosini, E.; Riva, M.; De Sensi, E.; Luppi, E.; Messina, C.; Caleffi, E.; Pisani, F.; et al. Reassessment of the NF1 variants of unknown significance found during the 20-year activity of a genetics diagnostic laboratory. Eur. J. Med. Genet. 2023, 66, 104847. [Google Scholar] [CrossRef]
- FDA. FDA Warning: Avoid Using Bone Growth Products in Children; FDA: Silver Spring, MD, USA, 2017. [Google Scholar]
- Sharma, R.; Wu, X.; Rhodes, S.D.; Chen, S.; He, Y.; Yuan, J.; Li, J.; Yang, X.; Li, X.; Jiang, L.; et al. Hyperactive Ras/MAPK signaling is critical for tibial nonunion fracture in neurofibromin-deficient mice. Hum. Mol. Genet. 2013, 22, 4818–4828. [Google Scholar] [CrossRef]
- Carlier, A.; Vasilevich, A.; Marechal, M.; de Boer, J.; Geris, L. In silico clinical trials for pediatric orphan diseases. Sci. Rep. 2018, 8, 2465. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y.; Zhu, K.; Li, J.; Guan, Y.; He, X.; Jin, X.; Bai, G.; Hu, L. Clinical characteristics and in silico analysis of congenital pseudarthrosis of the tibia combined with neurofibromatosis type 1 caused by a novel NF1 mutation. Front. Genet. 2022, 13, 991314. [Google Scholar] [CrossRef]
- Bettegowda, C.; Upadhayaya, M.; Evans, D.G.; Kim, A.; Mathios, D.; Hanemann, C.O.; Collaboration, R.E.I. Genotype-Phenotype Correlations in Neurofibromatosis and Their Potential Clinical Use. Neurology 2021, 97, S91–S98. [Google Scholar] [CrossRef]
- Upadhyaya, M.; Huson, S.M.; Davies, M.; Thomas, N.; Chuzhanova, N.; Giovannini, S.; Evans, D.G.; Howard, E.; Kerr, B.; Griffiths, S.; et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): Evidence of a clinically significant NF1 genotype-phenotype correlation. Am. J. Hum. Genet. 2007, 80, 140–151. [Google Scholar] [CrossRef]
- Pinna, V.; Lanari, V.; Daniele, P.; Consoli, F.; Agolini, E.; Margiotti, K.; Bottillo, I.; Torrente, I.; Bruselles, A.; Fusilli, C.; et al. p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur. J. Hum. Genet. 2015, 23, 1068–1071. [Google Scholar] [CrossRef]
- Kehrer-Sawatzki, H.; Mautner, V.F.; Cooper, D.N. Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum. Genet. 2017, 136, 349–376. [Google Scholar] [CrossRef]
- Futagawa, M.; Okazaki, T.; Nakata, E.; Fukano, C.; Osumi, R.; Kato, F.; Urakawa, Y.; Yamamoto, H.; Ozaki, T.; Hirasawa, A. Genotypes and phenotypes of neurofibromatosis type 1 patients in Japan: A Hereditary Tumor Cohort Study. Hum. Genome Var. 2024, 11, 42. [Google Scholar] [CrossRef]
- Ruggieri, M.; Polizzi, A.; Spalice, A.; Salpietro, V.; Caltabiano, R.; D’Orazi, V.; Pavone, P.; Pirrone, C.; Magro, G.; Platania, N.; et al. The natural history of spinal neurofibromatosis: A critical review of clinical and genetic features. Clin. Genet. 2015, 87, 401–410. [Google Scholar] [CrossRef]
- Napolitano, F.; Dell’Aquila, M.; Terracciano, C.; Franzese, G.; Gentile, M.T.; Piluso, G.; Santoro, C.; Colavito, D.; Patane, A.; De Blasiis, P.; et al. Genotype-Phenotype Correlations in Neurofibromatosis Type 1: Identification of Novel and Recurrent NF1 Gene Variants and Correlations with Neurocognitive Phenotype. Genes 2022, 13, 1130. [Google Scholar] [CrossRef]
- Poplin, R.; Chang, P.C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef]
- Luo, R.; Sedlazeck, F.J.; Lam, T.-W.; Schatz, M.C. A multi-task convolutional deep neural network for variant calling in single molecule sequencing. Nat. Commun. 2019, 10, 998. [Google Scholar] [CrossRef]
- Boza, V.; Brejova, B.; Vinar, T. DeepNano: Deep recurrent neural networks for base calling in MinION nanopore reads. PLoS ONE 2017, 12, e0178751. [Google Scholar] [CrossRef]
- Anzar, I.; Sverchkova, A.; Stratford, R.; Clancy, T. NeoMutate: An ensemble machine learning framework for the prediction of somatic mutations in cancer. BMC Med. Genom. 2019, 12, 63. [Google Scholar] [CrossRef]
- Spinella, J.F.; Mehanna, P.; Vidal, R.; Saillour, V.; Cassart, P.; Richer, C.; Ouimet, M.; Healy, J.; Sinnett, D. SNooPer: A machine learning-based method for somatic variant identification from low-pass next-generation sequencing. BMC Genom. 2016, 17, 912. [Google Scholar] [CrossRef]
- Ravasio, V.; Ritelli, M.; Legati, A.; Giacopuzzi, E. GARFIELD-NGS: Genomic vARiants FIltering by dEep Learning moDels in NGS. Bioinformatics 2018, 34, 3038–3040. [Google Scholar] [CrossRef]
- Singh, A.; Bhatia, P. Intelli-NGS: Intelligent NGS, a deep neural network-based artificial intelligence to delineate good and bad variant calls from IonTorrent sequencer data. bioRxiv 2019. [Google Scholar] [CrossRef]
- Quang, D.; Chen, Y.; Xie, X. DANN: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef]
- Gurovich, Y.; Hanani, Y.; Bar, O.; Nadav, G.; Fleischer, N.; Gelbman, D.; Basel-Salmon, L.; Krawitz, P.M.; Kamphausen, S.B.; Zenker, M.; et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat. Med. 2019, 25, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Boudellioua, I.; Kulmanov, M.; Schofield, P.N.; Gkoutos, G.V.; Hoehndorf, R. DeepPVP: Phenotype-based prioritization of causative variants using deep learning. BMC Bioinform. 2019, 20, 65. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhao, K.; Bustamante, C.D.; Ma, X.; Wong, W.H. Xrare: A machine learning method jointly modeling phenotypes and genetic evidence for rare disease diagnosis. Genet. Med. 2019, 21, 2126–2134. [Google Scholar] [CrossRef]
- Yang, Z.; Liu, X.; Xu, H.; Teschendorff, A.E.; Xu, L.; Li, J.; Fu, M.; Liu, J.; Zhou, H.; Wang, Y.; et al. Integrative analysis of genomic and epigenomic regulation reveals miRNA mediated tumor heterogeneity and immune evasion in lower grade glioma. Commun. Biol. 2024, 7, 824. [Google Scholar] [CrossRef]
- Yin, M.; Feng, C.; Yu, Z.; Zhang, Y.; Li, Y.; Wang, X.; Song, C.; Guo, M.; Li, C. sc2GWAS: A comprehensive platform linking single cell and GWAS traits of human. Nucleic Acids Res. 2025, 53, D1151–D1161. [Google Scholar] [CrossRef]
- Xing, Y.; Yang, K.; Lu, A.; Mackie, K.; Guo, F. Sensors and Devices Guided by Artificial Intelligence for Personalized Pain Medicine. Cyborg Bionic Syst. 2024, 5, 0160. [Google Scholar] [CrossRef]
- Zhou, C.; Kuang, M.; Tao, Y.; Wang, J.; Luo, Y.; Fu, Y.; Chen, Z.; Liu, Y.; Li, Z.; Wu, W.; et al. Nynrin preserves hematopoietic stem cell function by inhibiting the mitochondrial permeability transition pore opening. Cell Stem Cell 2024, 31, 1359–1375.e8. [Google Scholar] [CrossRef]
- Zhou, X.; Li, H.; Xie, Z. METTL3-modified exosomes from adipose-derived stem cells enhance the proliferation and migration of dermal fibroblasts by mediating m6A modification of CCNB1 mRNA. Arch. Dermatol. Res. 2025, 317, 418. [Google Scholar] [CrossRef]
- Sharo, A.G.; Zou, Y.; Adhikari, A.N.; Brenner, S.E. ClinVar and HGMD genomic variant classification accuracy has improved over time, as measured by implied disease burden. Genome Med. 2023, 15, 51. [Google Scholar] [CrossRef]
- Dawood, M.; Fayer, S.; Pendyala, S.; Post, M.; Kalra, D.; Patterson, K.; Venner, E.; Muffley, L.A.; Fowler, D.M.; Rubin, A.F.; et al. Using multiplexed functional data to reduce variant classification inequities in underrepresented populations. Genome Med. 2024, 16, 143. [Google Scholar] [CrossRef]
- Franklin by Genoox. Available online: https://franklin.genoox.com/about-franklin (accessed on 30 April 2025).
- Einhorn, Y.; Einhorn, M.; Kurolap, A.; Steinberg, D.; Mory, A.; Bazak, L.; Paperna, T.; Grinshpun-Cohen, J.; Basel-Salmon, L.; Weiss, K.; et al. Community data-driven approach to identify pathogenic founder variants for pan-ethnic carrier screening panels. Hum. Genom. 2023, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.J.; Tang, Y.; Sun, Y.B.; Yang, T.L.; Yan, C.; Liu, H.; Liu, J.; Huang, J.N.; Wang, M.H.; Yao, Z.W.; et al. A multicenter study of neurofibromatosis type 1 utilizing deep learning for whole body tumor identification. NPJ Digit. Med. 2025, 8, 56. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Variant Interpretation and Pathogenicity Prediction | |
| AI Tool | Function/Description |
| SpliceAI [22] | Deep learning model that predicts splice site disruptions across the entire transcript, enabling accurate detection of both canonical and non-canonical splice-altering variants in the NF1 gene. |
| REVEL [19,29] | Ensemble machine learning tool that combines scores from multiple individual predictors (e.g., SIFT, PolyPhen-2) to improve the classification of rare missense variants as likely benign or pathogenic. |
| VEST3 [29,30] | Supervised learning algorithm trained on known pathogenic and benign variants; uses sequence conservation, protein features, and structural data to predict functional impact of NF1 missense mutations. |
| ClinPred [28,29] | Machine learning classifier trained on ClinVar data; integrates multiple features including conservation, protein annotations, and clinical evidence to assess variant pathogenicity. |
| PredictSNP2 [31] | Consensus-based predictor that merges results from several established tools (e.g., SNAP, PANTHER, PhD-SNP) to enhance reliability in predicting the functional consequences of NF1 missense variants. |
| Align-GVGD [31] | Combines evolutionary conservation and biochemical properties to assess the functional impact of amino acid substitutions in NF1, particularly useful in cysteine mutation evaluation. |
| RENOVO-NF1 [41] | NF1-specific random forest model that calculates a Pathogenicity Likelihood Score (PLS) and effectively reclassifies NF1 missense VUS into likely pathogenic or benign with high accuracy. |
| DITTO [37] | Advanced AI model that integrates transcriptomic, proteomic, and structural dynamics data to evaluate the functional effects of NF1 mutations, including protein conformation-specific impacts. |
| SAAFEC-seq [37] | Gradient boosting-based model estimating protein stability changes (ΔΔG) using sequence-derived features to assess potential pathogenic effects of NF1 mutations. |
| Protein Structure and Stability Prediction | |
| AI Tool | Function/Description |
| AlphaFold3 [31,35] | Deep learning tool for predicting 3D protein structures at high resolution; used to visualize and assess how NF1 mutations affect neurofibromin folding and domain architecture. |
| iStable [31] | Integrates predictions from iMutant 2.0 and MUpro to estimate mutation-induced changes in protein stability (ΔΔG), helping identify destabilizing NF1 variants. |
| iMutant 2.0 [31] | SVM-based predictor for estimating the impact of single-point mutations on protein stability. |
| MUpro [31] | Combines SVM and neural networks to predict whether a mutation increases or decreases protein stability in NF1. |
| Tumor Classification | |
| AI Tool | Function/Description |
| GENIE-NF-AI [39] | Deep learning model based on a liquid neural network (LSTM) trained on gene expression data to classify NF1-associated tumors with high accuracy. It integrates black-box predictive performance with glass-box interpretability—using explainable AI layers to clarify how gene features contribute to classification, thus enhancing clinical trust and transparency. |
| Therapeutic Prediction | |
| AI Tool | Function/Description |
| In Silico AI Tools [46] | Machine learning framework used in virtual clinical trials for NF1-related CPT; includes random forest for response prediction, biomarker discovery, and patient stratification based on simulated biological outcomes. |
| AI Advantage | Description | Example/Application in NF1 |
|---|---|---|
| Enhanced Variant Interpretation | AI reduces uncertainty in classifying missense mutations and VUS. | Tools like REVEL, VEST3, and RENOVO-NF1 improve confidence in variant classification, aiding early diagnosis and risk assessment [29,41]. |
| Accurate Structural Impact Prediction | AI-powered structural models predict how mutations affect neurofibromin conformation. | AlphaFold3 and DITTO reveal stability changes in different protein states, offering insights for targeted therapies [31,35,37]. |
| Rapid Analysis of Big Genomic Data | AI accelerates processing of sequencing datasets, prioritizing clinically relevant variants. | In silico tools rapidly stratify patient data (e.g., CPT models in virtual trials), reducing diagnostic delays [46]. |
| Integration of Multi-Omics Data | AI can unify genomic, transcriptomic, and proteomic information for comprehensive variant assessment. | DITTO integrates transcriptomic and structural dynamics to model protein behavior across conformations [37]. |
| Support for Clinical Decision-Making | AI enhances diagnostic precision and treatment planning by reducing ambiguity. | GENIE-NF-AI and RENOVO-NF1 assist in tumor classification and VUS reclassification, guiding early interventions [39,41]. |
| Ethical Therapeutic Exploration | AI enables virtual clinical trials in populations where real trials are ethically challenging. | In silico BMP therapy trials for NF1-CPT model treatment outcomes in children without physical risk [46]. |
| Challenge | AI Limitation | Proposed Mitigation Strategy |
|---|---|---|
| Data Representation Bias | AI tools trained on databases like ClinVar and HGMD often reflect Eurocentric variant data, reducing performance on variants common in non-European populations [72]. | Promote use of diverse datasets and platforms like Franklin; integrate community-contributed variant data for broader ancestry coverage [74]. |
| Limited Generalizability | AI models may fail on novel or ultra-rare variants due to lack of similar examples in training data [72]. | Continually retrain models with updated real-world clinical data and include synthetic data from simulated environments where appropriate [74]. |
| Lack of Functional Validation | AI predictions often lack biological validation, reducing clinical trust. | Use multiplexed assays of variant effects (MAVEs) and encourage AI–wet lab partnerships to validate predictions [73]. |
| Missense Variant Focus | Most tools are optimized for missense mutations and lack support for intronic, splicing, or structural variants. | Incorporate tools like SpliceAI to cover splicing and regulatory regions [22]. |
| Interpretability and Clinical Trust | Black-box models limit clinical adoption due to poor transparency in decision-making [39]. | Use explainable AI (e.g., GENIE-NF-AI’s glass-box overlay) to make model logic transparent for clinicians [39]. |
| Regulatory and Integration Barriers | Many AI tools are not validated for clinical use, delaying integration into routine diagnostics [39,41]. | Develop standards for AI validation and interoperability in genomics workflows, aligned with ACMG frameworks. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grech, V.S.; Lotsaris, K.; Touma, T.E.; Kefala, V.; Rallis, E. The Role of Artificial Intelligence in Identifying NF1 Gene Variants and Improving Diagnosis. Genes 2025, 16, 560. https://doi.org/10.3390/genes16050560
Grech VS, Lotsaris K, Touma TE, Kefala V, Rallis E. The Role of Artificial Intelligence in Identifying NF1 Gene Variants and Improving Diagnosis. Genes. 2025; 16(5):560. https://doi.org/10.3390/genes16050560
Chicago/Turabian StyleGrech, Vasiliki Sofia, Kleomenis Lotsaris, Theano Eirini Touma, Vassiliki Kefala, and Efstathios Rallis. 2025. "The Role of Artificial Intelligence in Identifying NF1 Gene Variants and Improving Diagnosis" Genes 16, no. 5: 560. https://doi.org/10.3390/genes16050560
APA StyleGrech, V. S., Lotsaris, K., Touma, T. E., Kefala, V., & Rallis, E. (2025). The Role of Artificial Intelligence in Identifying NF1 Gene Variants and Improving Diagnosis. Genes, 16(5), 560. https://doi.org/10.3390/genes16050560