
A Rare Homozygous AP4S1 Variant in Rwandan Siblings with Autosomal Recessive Hereditary Spastic Paraplegia Type 52 (SPG52)

 ,
,  , , , , , , , , ,
, , , , , , , , ,
Abstract
1. Introduction
2. Materials and Methods
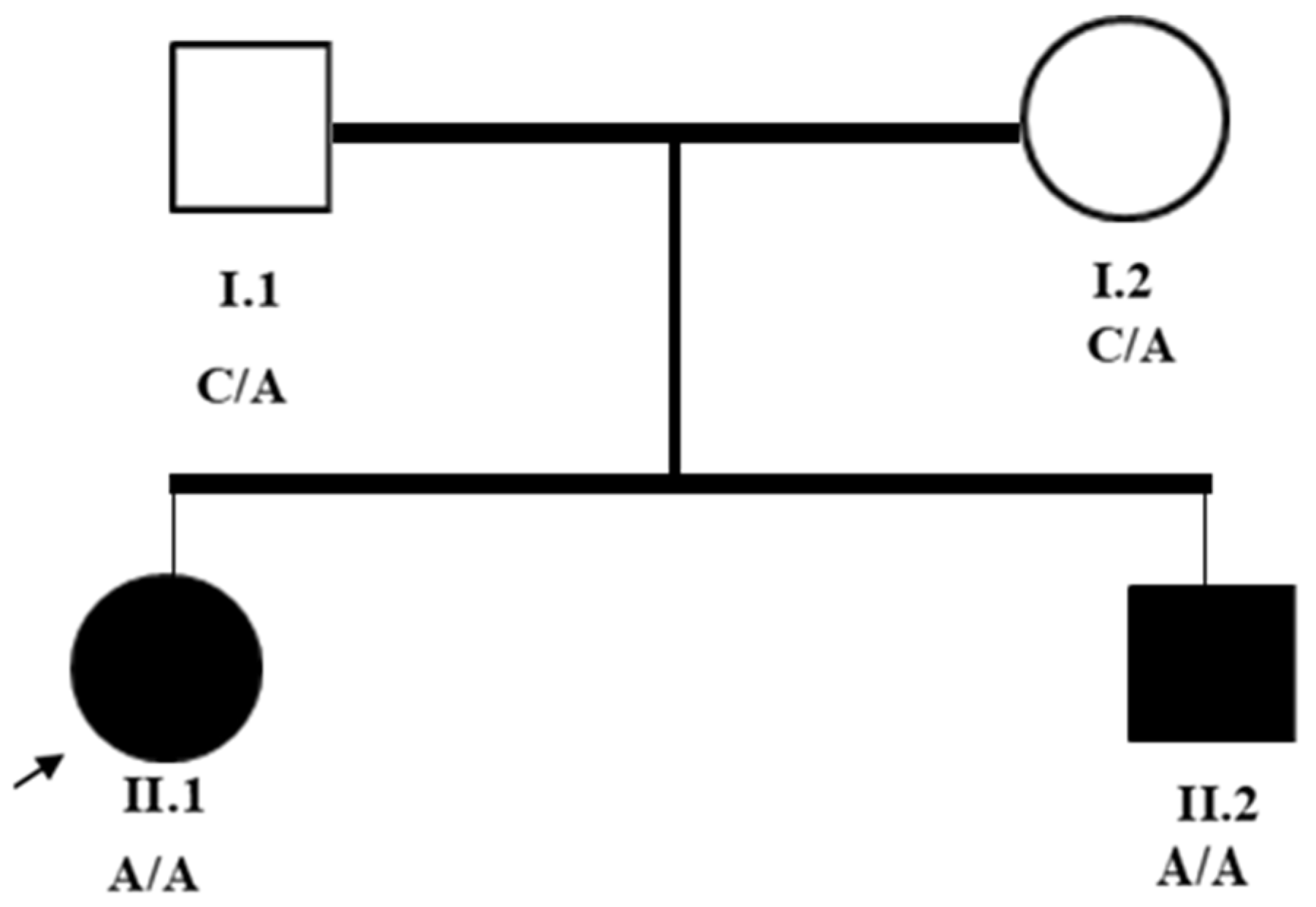
2.1. Subjects and Clinical Assessment
2.2. Imaging and Molecular Investigations
3. Results
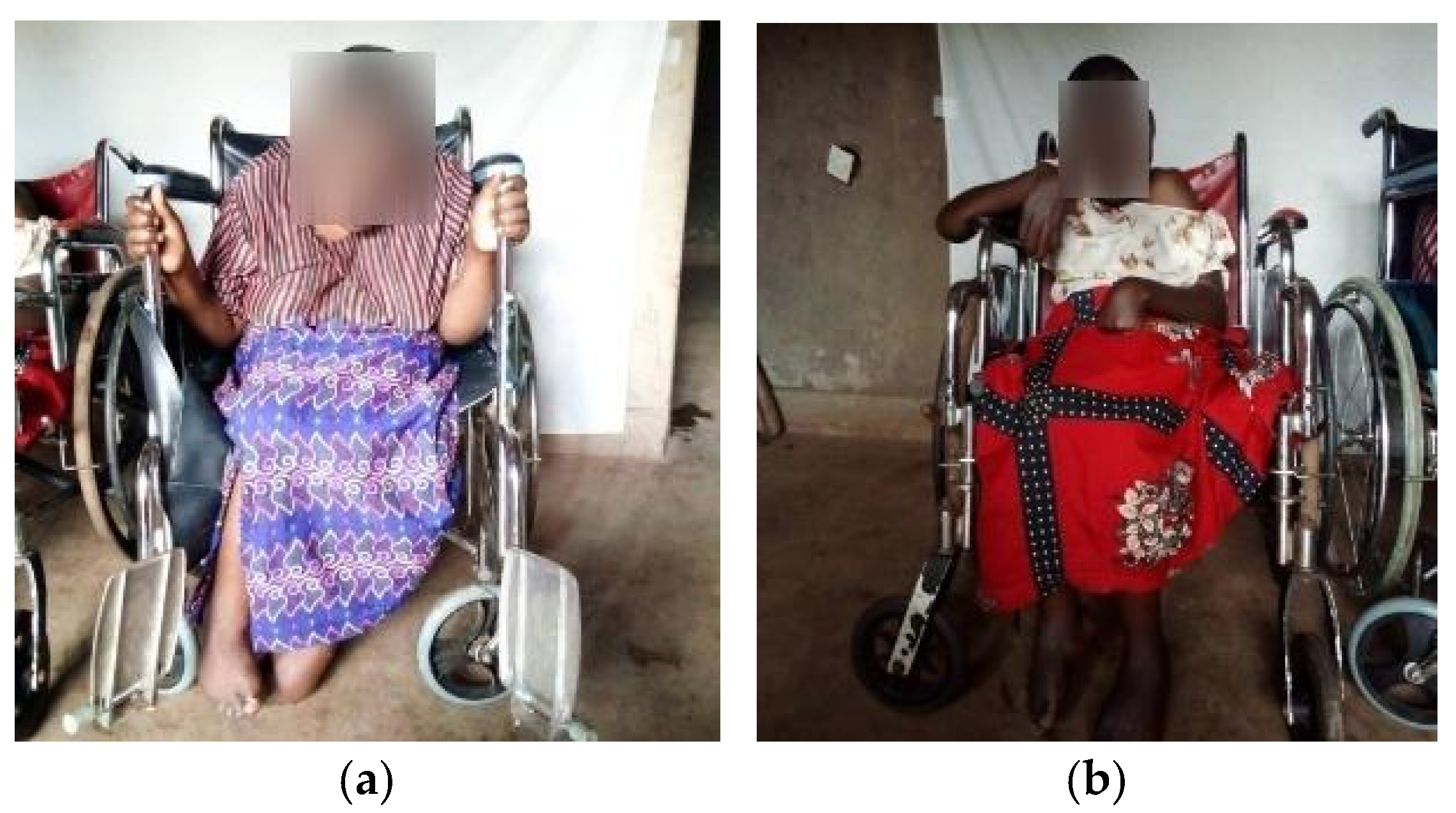
3.1. Clinical Assessments
3.2. Imaging Analysis
3.3. Exome Sequencing Analysis
3.4. Pathogenicity Interpretation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| SPG | Spastic Paraplegia |
| HSP | Hereditary Spastic Paraplegia |
| MRI | Magnetic Resonance Imaging |
| WES | Whole-Exome Sequencing |
| NM | Nuclear mRNA |
| AP | Adaptor Protein Complex |
| ACMG | American College of Medical Genetics and Genomics |
| Hg19 | Human Genome 19 |
| RNA-Seq | RNA Sequencing |
| AP4B1 | β-1 Subunit, Adaptor Protein Complex 4 |
| AP4E1 | Epsilon-1 Subunit, Adaptor Protein Complex 4 |
| AP4M1 | Mu-1 Subunit, Adaptor Protein Complex 4 |
| AP4S1 | Sigma-1 Subunit, Adaptor Protein Complex 4 |
| ATN1 | Atrophin 1 |
| ATXN1 | Ataxin 1 |
| ATXN10 | Ataxin 10 |
| ATXN2 | Ataxin 2 |
| ATXN3 | Ataxin 3 |
| ATXN7 | Ataxin 7 |
| ATXN8OS | Ataxin 8 Opposite Strand |
| BEAN1 | Brain-Expressed Associated with NEDD4 (Neural Precursor Cell-Expressed Developmentally Down-Regulated 4), 1 |
| CACNA1A | Calcium Voltage-Gated Channel Subunit Alpha1 A |
| CNV | Copy Number Variation |
| FXN | Frataxin |
| GRCh37 | Genome Reference Consortium Human Build 37 |
| NOP56 | NOP56 Ribonucleoprotein |
| PCR | Polymerase Chain Reaction |
| PPP2R2B | Protein Phosphatase 2 Regulatory unit B β |
| RPA | Repeat-Primed Assay |
| SPG47 | Spastic Paraplegia 47 |
| SPG50 | Spastic Paraplegia 50 |
| SPG51 | Spastic Paraplegia 51 |
| SPG52 | Spastic Paraplegia 52 |
| TBP | TATA-Box-Binding Protein |
| gnomAD | Genome Aggregation Database |
| PS3 | Pathogenic Strong 3 |
| PM2 | Pathogenic Moderate 2 |
| PP3 | Pathogenic Supporting 3 |
| PP5 | Pathogenic Supporting 5 |
| HiSeq | High-Throughput Sequencing System |
| NextSeq | Next-Generation Sequencing System |
| Ada | AdaBoost Score |
| RF | Random Forest Score |
References
- Brasch, J.; Shribman, S.; Reid, E.; Crosby, A.H.; Houlden, H.; Warner, T.T. Hereditary spastic paraplegia: From diagnosis to emerging therapeutic approaches. Lancet Neurol. 2019, 18, 1136–1146. [Google Scholar] [CrossRef]
- Meyyazhagan, A.; Orlacchio, A. Hereditary Spastic Paraplegia: An Update. Int. J. Mol. Sci. 2022, 23, 1697. [Google Scholar] [CrossRef] [PubMed]
- Bellofatto, M.; De Michele, G.; Iovino, A.; Filla, A.; Santorelli, F.M. Management of hereditary spastic paraplegia: A systematic review of the literature. Front. Neurol. 2019, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Schüle, R.; Wiethoff, S.; Martus, P.; Karle, K.N.; Otto, S.; Klebe, S.; Klimpe, S.; Gallenmüller, C.; Kurzwelly, D.; Henkel, D.; et al. Hereditary spastic paraplegia: Clinicogenetic lessons from 608 patients. Ann. Neurol. 2016, 79, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Tessa, A.; Battini, R.; Rubegni, A.; Storti, E.; Marini, C.; Galatolo, D.; Pasquariello, R.; Barghigiani, M.; Sgherri, G.; Chimenti, I.; et al. Identification of mutations in AP4S1/SPG52 through next generation sequencing in three families. Eur. J. Neurol. 2016, 23, 1580–1587. [Google Scholar] [CrossRef] [PubMed]
- Moreno-De-Luca, A.; Helmers, S.L.; Mao, H.; Burns, T.G.; Melton, A.M.A.; Schmidt, K.R.; Fernhoff, P.M.; Ledbetter, D.H.; Martin, C.L. Adaptor protein complex-4 (AP-4) deficiency causes a novel autosomal recessive cerebral palsy syndrome with microcephaly and intellectual disability. J. Med. Genet. 2011, 48, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Trummer, B.; Haubenberger, D.; Blackstone, C. Clinical Trial Designs and Measures in Hereditary Spastic Paraplegias. Front. Neurol. 2018, 9, 1017. [Google Scholar] [CrossRef] [PubMed]
- ClinVar. NM_001128126.3(AP4S1):c.295-3C>A. Accession: RCV001089600.1. National Center for Biotechnology Information Archives. Available online: https://www.ncbi.nlm.nih.gov/clinvar/RCV001089600.1/ (accessed on 21 April 2025).
- McCullough, C.G.; Szelinger, S.; Belnap, N.; Ramsey, K.; Schrauwen, I.; Claasen, A.M.; Burke, L.W.; Siniard, A.L.; Huentelman, M.J.; Narayanan, V.; et al. Utilizing RNA and outlier analysis to identify an intronic splice-altering variant in AP4S1 in a sibling pair with progressive spastic paraplegia. Hum. Mutat. 2020, 41, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The Global Epidemiology of Hereditary Ataxia and Spastic Paraplegia: A Systematic Review of Prevalence Studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Bouwkamp, C.G.; Afawi, Z.; Fattal-Valevski, A.; Krabbendam, I.E.; Rivetti, S.; Masalha, R.; Quadri, M.; Breedveld, G.J.; Mendel, H.; Tailakh, M.A.; et al. ACO2 homozygous missense mutation associated with complicated hereditary spastic paraplegia. Neurol. Genet. 2018, 4, e223. [Google Scholar] [CrossRef]
- Carmona, S.; Marecos, C.; Amorim, M.; Ferreira, A.C.; Conceição, C.; Brás, J.; Duarte, S.T.; Guerreiro, R. Splice-site mutation in AP4S1 causing spastic paraplegia type 52 with polymicrogyria: A case report. Neurol. Genet. 2018, 4, e273. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alecu, J.; Schierbaum, L.; Ebrahimi-Fakhari, D. AP-4-Associated Hereditary Spastic Paraplegia. In GeneReviews®; University of Washington: Seattle, WA, USA, 2018. [Google Scholar]
- Mahungu, A.C.; Steyn, E.; Floudiotis, N.; Wilson, L.A.; Vandrovcova, J.; Reilly, M.M.; Record, C.J.; Benatar, M.; Wu, G.; Raga, S.; et al. The mutational profile in a South African cohort with inherited neuropathies and spastic paraplegia. Front. Neurol. 2023, 14, 1239725. [Google Scholar] [CrossRef] [PubMed]
- CENTOGENE. Genetic Testing Offer for Neurology; CENTOGENE: Rostock, Germany, 2025. [Google Scholar]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. gnomAD Variant: 14-31080570-C-A. Genome Aggregation Database (gnomAD) v4. Broad Institute. 2024. Available online: https://gnomad.broadinstitute.org/variant/14-31080570-C-A?dataset=gnomad_r4 (accessed on 23 April 2015).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Teinert, J.; Behne, R.; Wimmer, M.; D’Amore, A.; Eberhardt, K.; Brechmann, B.; Ziegler, M.; Jensen, D.M.; Nagabhyrava, P.; et al. Adaptor protein complex 4-associated hereditary spastic paraplegia clinical, molecular and imaging spectrum. Brain 2020, 143, 2929–2944, Erratum in: Brain 2021, 144, e33. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Morales, J.; Pujar, S.; Loveland, J.E.; Astashyn, A.; Bennett, R.; Berry, A.; Cox, E.; Davidson, C.; Ermolaeva, O.; Farrell, C.M.; et al. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature 2022, 604, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Fink, J.K. Hereditary spastic paraplegia: Clinico-pathologic features and emerging molecular mechanisms. Acta Neuropathol. 2013, 126, 307–328. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Goldman, J.S. Predictive genetic counseling for neurodegenerative diseases: Past, present, and future. Cold Spring Harb. Perspect. Med. 2020, 10, a036525. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Gene Symbol | Gene Name | Associated Disorder(s) | Inheritance Mode |
|---|---|---|---|
| ATN1 | Atrophin 1 | Dentatorubral–Pallidoluysian Atrophy (DRPLA) | Autosomal Dominant |
| ATXN1 | Ataxin 1 | Spinocerebellar Ataxia Type 1 (SCA1) | Autosomal Dominant |
| ATXN2 | Ataxin 2 | Spinocerebellar Ataxia Type 2 (SCA2), ALS Modifier | Autosomal Dominant |
| ATXN3 | Ataxin 3 | Spinocerebellar Ataxia Type 3 (SCA3, Machado–Joseph Disease) | Autosomal Dominant |
| ATXN7 | Ataxin 7 | Spinocerebellar Ataxia Type 7 (SCA7) | Autosomal Dominant |
| ATXN8OS | Ataxin 8 Opposite Strand | Spinocerebellar Ataxia Type 8 (SCA8) | Autosomal Dominant (Incomplete Penetrance) |
| ATXN10 | Ataxin 10 | Spinocerebellar Ataxia Type 10 (SCA10) | Autosomal Dominant |
| BEAN1 | Brain-Expressed and Associated with NEDD4, 1 | Spastic Paraplegia 75 | Autosomal Recessive |
| CACNA1A | Calcium Voltage-Gated Channel Subunit Alpha1 A | Episodic Ataxia, SCA6, Familial Hemiplegic Migraine | Autosomal Dominant |
| FXN | Frataxin | Friedreich Ataxia | Autosomal Recessive |
| NOP56 | NOP56 Ribonucleoprotein | Spinocerebellar Ataxia Type 36 (SCA36) | Autosomal Dominant |
| PPP2R2B | Protein Phosphatase 2 Regulatory Subunit B β | Spinocerebellar Ataxia Type 12 (SCA12) | Autosomal Dominant |
| TBP | TATA-Box-Binding Protein | Spinocerebellar Ataxia Type 17 (SCA17) | Autosomal Dominant |
| AP4S1 | Adaptor-Related Protein Complex 4 Subunit Sigma 1 | Spastic Paraplegia 52 (SPG52) | Autosomal Recessive |
| Criterion Code | Description | Strength * |
|---|---|---|
| Pathogenic Evidence | ||
| PS3 | Functional studies are supportive of a damaging effect | Strong |
| PM2 | Absent or very low frequency in large population databases | Moderate |
| PP3 | Multiple lines of computational evidence support a deleterious effect | Supporting |
| PP5 | A variant reported in reputable sources as pathogenic | Supporting |
| Feature | European Cases (Tessa et al., 2021; Abou Jamra et al., 2011) [5] | Rwandan Siblings (This Study) |
|---|---|---|
| Age of Onset | 6 months–3 years (neonatal hypotonia → childhood spasticity) | 3–5 years (delayed motor milestones) |
| Severity | Moderate–severe (wheelchair dependence common) | Severe (wheelchair-dependent by adulthood) |
| Seizures | Reported in ~50% of cases | Present (managed with phenobarbital) |
| Speech Delay | Frequent (mutism in some) | Present (dysarthria) |
| Similar response to therapy | ||
| Treatment Response | Partial improvement with physiotherapy/muscle relaxants |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niyoyita, S.; Uwibambe, E.; Ndinkabandi, J.; Sesonga, P.; Niyongere, J.B.; Tuyishimire, B.; Urugwiro, A.; Rwamatwara, A.; Isingizwe, G.; Mutamuliza, J.; et al. A Rare Homozygous AP4S1 Variant in Rwandan Siblings with Autosomal Recessive Hereditary Spastic Paraplegia Type 52 (SPG52). Genes 2025, 16, 542. https://doi.org/10.3390/genes16050542
Niyoyita S, Uwibambe E, Ndinkabandi J, Sesonga P, Niyongere JB, Tuyishimire B, Urugwiro A, Rwamatwara A, Isingizwe G, Mutamuliza J, et al. A Rare Homozygous AP4S1 Variant in Rwandan Siblings with Autosomal Recessive Hereditary Spastic Paraplegia Type 52 (SPG52). Genes. 2025; 16(5):542. https://doi.org/10.3390/genes16050542
Chicago/Turabian StyleNiyoyita, Sylvine, Esther Uwibambe, Janvier Ndinkabandi, Placide Sesonga, Josse Belladone Niyongere, Benjamin Tuyishimire, Adelaide Urugwiro, Alype Rwamatwara, Gisèle Isingizwe, Janvière Mutamuliza, and et al. 2025. "A Rare Homozygous AP4S1 Variant in Rwandan Siblings with Autosomal Recessive Hereditary Spastic Paraplegia Type 52 (SPG52)" Genes 16, no. 5: 542. https://doi.org/10.3390/genes16050542
APA StyleNiyoyita, S., Uwibambe, E., Ndinkabandi, J., Sesonga, P., Niyongere, J. B., Tuyishimire, B., Urugwiro, A., Rwamatwara, A., Isingizwe, G., Mutamuliza, J., Nsanzabaganwa, C., Bukuru, J., Rutagarama, F., Mukaruziga, A., Karangwa, O., Ndatinya, A., Nsanzabera, M., Dukuze, N., & Mutesa, L. (2025). A Rare Homozygous AP4S1 Variant in Rwandan Siblings with Autosomal Recessive Hereditary Spastic Paraplegia Type 52 (SPG52). Genes, 16(5), 542. https://doi.org/10.3390/genes16050542