
Characterization of Extrachromosomal Circular DNA in Primary and Cisplatin-Resistant High-Grade Serous Ovarian Cancer

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Cell Counting Kit-8 Assay
2.3. Whole-Genome Sequencing, Data Preprocessing, and Alignment
2.4. Somatic Mutation Calling, Filtering, and Annotation
2.5. Mutational Signature Analysis of Single-Nucleotide Substitutions
2.6. Somatic Structural Variant Calling and Filtering
2.7. Copy Number Alterations Analysis
2.8. Detection of Complex Rearrangements
2.9. Circular DNA Sequencing
2.10. Identification of Unique and Shared eccDNAs
2.11. Identification of Unique Circular DNA Genes
2.12. RNA Sequencing Analysis
2.13. Statistical Analysis
3. Results
3.1. Establishment of Primary and Cisplatin-Resistant HGSOC Cell Lines
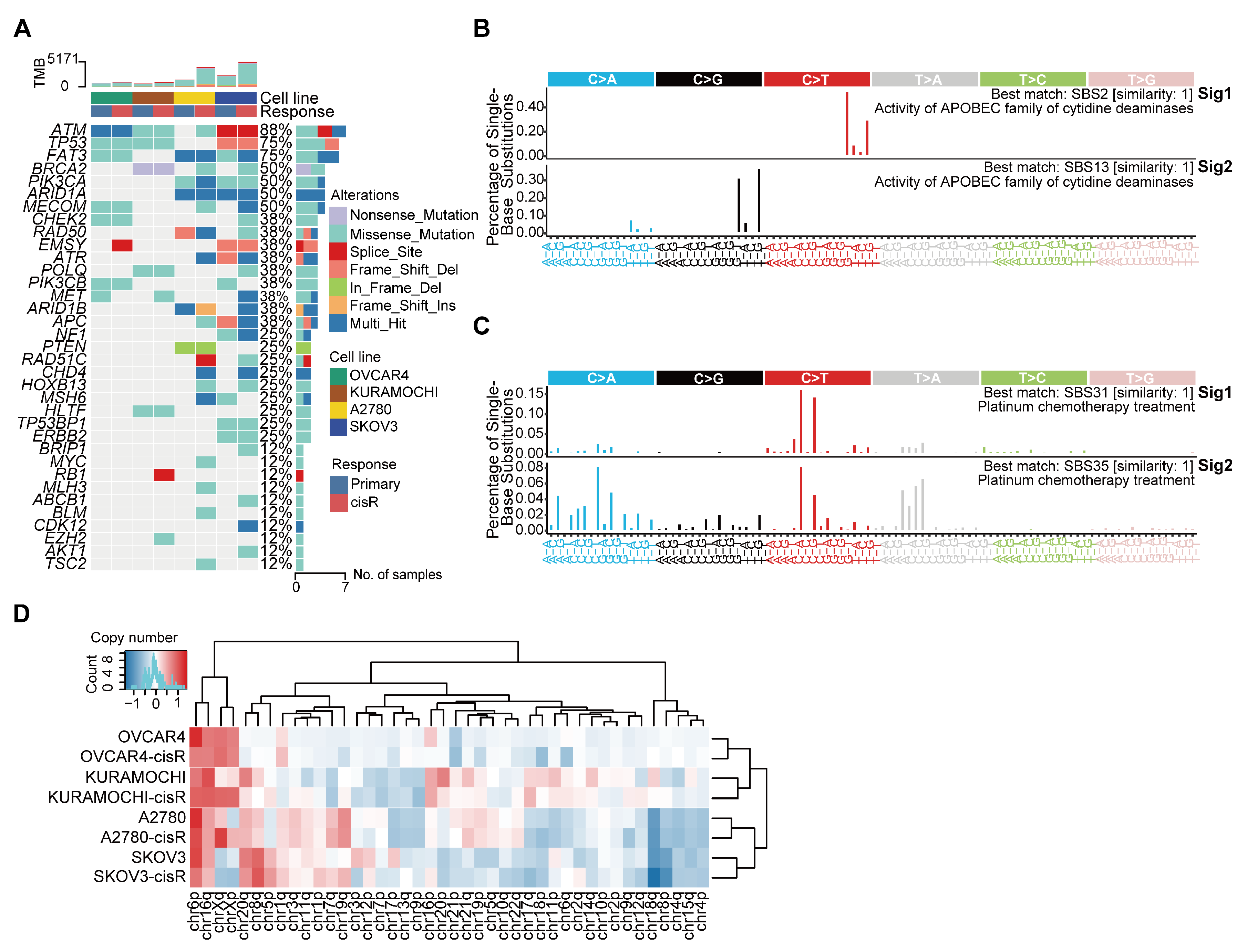
3.2. Genomic Features and Mutational Signatures in Primary and Cisplatin-Resistant HGSOC Cell Lines
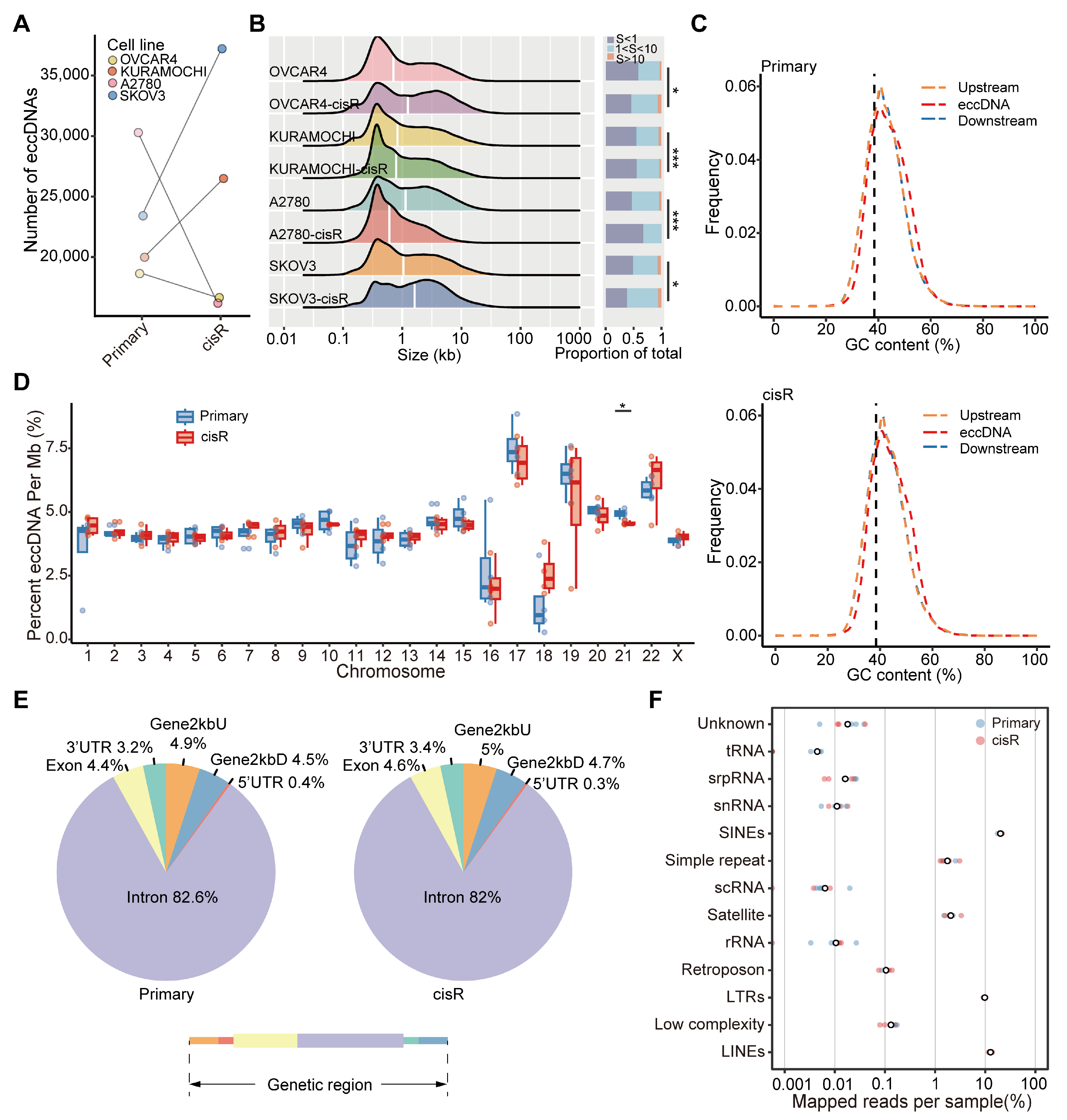
3.3. Characteristics of Large ecDNA in Primary and cisR HGSOC Cell Lines
3.4. Genome-Wide Detection and Distribution Features of Small eccDNAs in HGSOC Cell Lines
3.5. Differentially Expressed Gene Profiles and Functional Analysis in Primary and cisR Cell Lines
3.6. Differential Expression and Enrichment Analysis of eccDNA Encoding Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ovarian Cancer. Nat. Rev. Dis. Primers 2016, 2, 16062. [CrossRef] [PubMed]
- Go, R.S.; Adjei, A.A. Review of the Comparative Pharmacology and Clinical Activity of Cisplatin and Carboplatin. J. Clin. Oncol. 1999, 17, 409–422. [Google Scholar] [CrossRef]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole–Genome Characterization of Chemoresistant Ovarian Cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Wu, S.; Bafna, V.; Chang, H.Y.; Mischel, P.S. Extrachromosomal DNA: An Emerging Hallmark in Human Cancer. Annu. Rev. Pathol. 2022, 17, 367–386. [Google Scholar] [CrossRef]
- Ling, X.; Han, Y.; Meng, J.; Zhong, B.; Chen, J.; Zhang, H.; Qin, J.; Pang, J.; Liu, L. Small Extrachromosomal Circular DNA (eccDNA): Major Functions in Evolution and Cancer. Mol. Cancer 2021, 20, 113. [Google Scholar] [CrossRef]
- Liao, Z.; Jiang, W.; Ye, L.; Li, T.; Yu, X.; Liu, L. Classification of Extrachromosomal Circular DNA with a Focus on the Role of Extrachromosomal DNA (ecDNA) in Tumor Heterogeneity and Progression. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2020, 1874, 188392. [Google Scholar] [CrossRef]
- Nathanson, D.A.; Gini, B.; Mottahedeh, J.; Visnyei, K.; Koga, T.; Gomez, G.; Eskin, A.; Hwang, K.; Wang, J.; Masui, K.; et al. Targeted Therapy Resistance Mediated by Dynamic Regulation of Extrachromosomal Mutant EGFR DNA. Science 2014, 343, 72–76. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, M.; Djekidel, M.N.; Chen, H.; Liu, D.; Alt, F.W.; Zhang, Y. eccDNAs Are Apoptotic Products with High Innate Immunostimulatory Activity. Nature 2021, 599, 308–314. [Google Scholar] [CrossRef]
- Chen, Y.-A.; Shen, Y.-L.; Hsia, H.-Y.; Tiang, Y.-P.; Sung, T.-L.; Chen, L.-Y. Extrachromosomal Telomere Repeat DNA Is Linked to ALT Development via cGAS-STING DNA Sensing Pathway. Nat. Struct. Mol. Biol. 2017, 24, 1124–1131. [Google Scholar] [CrossRef]
- Møller, H.D.; Mohiyuddin, M.; Prada-Luengo, I.; Sailani, M.R.; Halling, J.F.; Plomgaard, P.; Maretty, L.; Hansen, A.J.; Snyder, M.P.; Pilegaard, H.; et al. Circular DNA Elements of Chromosomal Origin Are Common in Healthy Human Somatic Tissue. Nat. Commun. 2018, 9, 1069. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, T.; Shibata, Y.; Kumar, P.; Dillon, L.; Dutta, A. Small Extrachromosomal Circular DNAs, microDNA, Produce Short Regulatory RNAs That Suppress Gene Expression Independent of Canonical Promoters. Nucleic Acids Res. 2019, 47, 4586–4596. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Faust, G.G.; Hall, I.M. SAMBLASTER: Fast Duplicate Marking and Structural Variant Read Extraction. Bioinformatics 2014, 30, 2503–2505. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and Accurate Calling of Germline and Somatic Variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive Detection of Somatic Point Mutations in Impure and Heterogeneous Cancer Samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Talsania, K.; Shen, T.-W.; Chen, X.; Jaeger, E.; Li, Z.; Chen, Z.; Chen, W.; Tran, B.; Kusko, R.; Wang, L.; et al. Structural Variant Analysis of a Cancer Reference Cell Line Sample Using Multiple Sequencing Technologies. Genome Biol. 2022, 23, 255. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Islam, S.M.A.; Díaz-Gay, M.; Wu, Y.; Barnes, M.; Vangara, R.; Bergstrom, E.N.; He, Y.; Vella, M.; Wang, J.; Teague, J.W.; et al. Uncovering Novel Mutational Signatures by de Novo Extraction with SigProfilerExtractor. Cell Genom. 2022, 2, 100179. [Google Scholar] [CrossRef]
- Wala, J.A.; Bandopadhayay, P.; Greenwald, N.F.; O’Rourke, R.; Sharpe, T.; Stewart, C.; Schumacher, S.; Li, Y.; Weischenfeldt, J.; Yao, X.; et al. SvABA: Genome-Wide Detection of Structural Variants and Indels by Local Assembly. Genome Res. 2018, 28, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid Detection of Structural Variants and Indels for Germline and Cancer Sequencing Applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef] [PubMed]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stütz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural Variant Discovery by Integrated Paired-End and Split-Read Analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Seshan, V.E. FACETS: Allele-Specific Copy Number and Clonal Heterogeneity Analysis Tool for High-Throughput DNA Sequencing. Nucleic Acids Res. 2016, 44, e131. [Google Scholar] [CrossRef]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 Facilitates Sensitive and Confident Localization of the Targets of Focal Somatic Copy-Number Alteration in Human Cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef]
- Deshpande, V.; Luebeck, J.; Nguyen, N.-P.D.; Bakhtiari, M.; Turner, K.M.; Schwab, R.; Carter, H.; Mischel, P.S.; Bafna, V. Exploring the Landscape of Focal Amplifications in Cancer Using AmpliconArchitect. Nat. Commun. 2019, 10, 392. [Google Scholar] [CrossRef]
- Hadi, K.; Yao, X.; Behr, J.M.; Deshpande, A.; Xanthopoulakis, C.; Tian, H.; Kudman, S.; Rosiene, J.; Darmofal, M.; DeRose, J.; et al. Distinct Classes of Complex Structural Variation Uncovered across Thousands of Cancer Genome Graphs. Cell 2020, 183, 197–210.e32. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating Cell Lines as Tumour Models by Comparison of Genomic Profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.K.; Bashashati, A.; Anglesio, M.S.; Cochrane, D.R.; Grewal, D.S.; Ha, G.; McPherson, A.; Horlings, H.M.; Senz, J.; Prentice, L.M.; et al. Genomic Consequences of Aberrant DNA Repair Mechanisms Stratify Ovarian Cancer Histotypes. Nat. Genet. 2017, 49, 856–865. [Google Scholar] [CrossRef]
- Serebrenik, A.A.; Argyris, P.P.; Jarvis, M.C.; Brown, W.L.; Bazzaro, M.; Vogel, R.I.; Erickson, B.K.; Lee, S.-H.; Goergen, K.M.; Maurer, M.J.; et al. The DNA Cytosine Deaminase APOBEC3B Is a Molecular Determinant of Platinum Responsiveness in Clear Cell Ovarian Cancer. Clin. Cancer Res. 2020, 26, 3397–3407. [Google Scholar] [CrossRef]
- Cortés-Ciriano, I.; Lee, J.J.-K.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.-Z.; Pellman, D.S.; et al. Comprehensive Analysis of Chromothripsis in 2,658 Human Cancers Using Whole-Genome Sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef]
- Nguyen, D.D.; Hooper, W.F.; Liu, W.; Chu, T.R.; Geiger, H.; Shelton, J.M.; Shah, M.; Goldstein, Z.R.; Winterkorn, L.; Helland, A.; et al. The Interplay of Mutagenesis and ecDNA Shapes Urothelial Cancer Evolution. Nature 2024, 635, 219–228. [Google Scholar] [CrossRef]
- Yi, E.; Chamorro González, R.; Henssen, A.G.; Verhaak, R.G.W. Extrachromosomal DNA Amplifications in Cancer. Nat. Rev. Genet. 2022, 23, 760–771. [Google Scholar] [CrossRef]
- Huang, D.; Savage, S.R.; Calinawan, A.P.; Lin, C.; Zhang, B.; Wang, P.; Starr, T.K.; Birrer, M.J.; Paulovich, A.G. A Highly Annotated Database of Genes Associated with Platinum Resistance in Cancer. Oncogene 2021, 40, 6395–6405. [Google Scholar] [CrossRef]
- Cen, Y.; Fang, Y.; Ren, Y.; Hong, S.; Lu, W.; Xu, J. Global Characterization of Extrachromosomal Circular DNAs in Advanced High Grade Serous Ovarian Cancer. Cell Death Dis. 2022, 13, 342. [Google Scholar] [CrossRef]
- Yang, L.; Jia, R.; Ge, T.; Ge, S.; Zhuang, A.; Chai, P.; Fan, X. Extrachromosomal Circular DNA: Biogenesis, Structure, Functions and Diseases. Signal Transduct. Target. Ther. 2022, 7, 342. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Jiménez, F.; Movasati, A.; Brunner, S.R.; Nguyen, L.; Priestley, P.; Cuppen, E.; Van Hoeck, A. Pan-Cancer Whole-Genome Comparison of Primary and Metastatic Solid Tumours. Nature 2023, 618, 333–341. [Google Scholar] [CrossRef]
- Zhu, X.; Shen, H.; Yin, X.; Yang, M.; Wei, H.; Chen, Q.; Feng, F.; Liu, Y.; Xu, W.; Li, Y. Macrophages Derived Exosomes Deliver miR-223 to Epithelial Ovarian Cancer Cells to Elicit a Chemoresistant Phenotype. J. Exp. Clin. Cancer Res. 2019, 38, 81. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Chen, Y.; Zhang, F.; Liu, B.; Xie, C.; Song, Y. Encoding Gene RAB3B Exists in Linear Chromosomal and Circular Extrachromosomal DNA and Contributes to Cisplatin Resistance of Hypopharyngeal Squamous Cell Carcinoma via Inducing Autophagy. Cell Death Dis. 2022, 13, 171. [Google Scholar] [CrossRef] [PubMed]
- Helmsauer, K.; Valieva, M.E.; Ali, S.; Chamorro González, R.; Schöpflin, R.; Röefzaad, C.; Bei, Y.; Dorado Garcia, H.; Rodriguez-Fos, E.; Puiggròs, M.; et al. Enhancer Hijacking Determines Extrachromosomal Circular MYCN Amplicon Architecture in Neuroblastoma. Nat. Commun. 2020, 11, 5823. [Google Scholar] [CrossRef]
- Pietilä, E.A.; Gonzalez-Molina, J.; Moyano-Galceran, L.; Jamalzadeh, S.; Zhang, K.; Lehtinen, L.; Turunen, S.P.; Martins, T.A.; Gultekin, O.; Lamminen, T.; et al. Co-Evolution of Matrisome and Adaptive Adhesion Dynamics Drives Ovarian Cancer Chemoresistance. Nat. Commun. 2021, 12, 3904. [Google Scholar] [CrossRef]
- Liu, O.; Wang, C.; Wang, S.; Hu, Y.; Gou, R.; Dong, H.; Li, S.; Li, X.; Lin, B. Keratin 80 Regulated by miR-206/ETS1 Promotes Tumor Progression via the MEK/ERK Pathway in Ovarian Cancer. J. Cancer 2021, 12, 6835–6850. [Google Scholar] [CrossRef]
- Lin, J.; Fan, X.; Chen, J.; Xie, X.; Yu, H. Small Interfering RNA-Mediated Knockdown of KRT80 Suppresses Colorectal Cancer Proliferation. Exp. Ther. Med. 2020, 20, 176. [Google Scholar] [CrossRef]
- Shi, K.-H.; Xue, H.; Zhao, E.-H.; Xiao, L.-J.; Sun, H.-Z.; Zheng, H.-C. KRT80 Expression Works as a Biomarker and a Target for Differentiation in Gastric Cancer. Histol. Histopathol. 2024, 39, 117–130. [Google Scholar]
- Jiang, J.; Lu, J.; Feng, Y.; Zhao, Y.; Su, J.; Zeng, T.; Chen, Y.; Shen, K.; Jia, Y.; Lin, S. Identification of KRT80 as a Novel Prognostic and Predictive Biomarker of Human Lung Adenocarcinoma via Bioinformatics Approaches. Comb. Chem. High Throughput Screen. 2024; ahead of print. [Google Scholar]
- Yun, W.-J.; Li, J.; Yin, N.-C.; Zhang, C.-Y.; Cui, Z.-G.; Zhang, L.; Zheng, H.-C. The Facilitating Effects of KRT80 on Chemoresistance, Lipogenesis, and Invasion of Esophageal Cancer. Cancer Biol. Ther. 2024, 25, 2302162. [Google Scholar] [CrossRef]
- Wang, X.; Ye, X.; Chen, Y.; Lin, J. Mechanism of M2 Type Macrophage-Derived Extracellular Vesicles Regulating PD-L1 Expression via the MISP/IQGAP1 Axis in Hepatocellular Carcinoma Immunotherapy Resistance. Int. Immunopharmacol. 2023, 124, 110848. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ding, Z.; Peng, Y.; Pan, F.; Li, J.; Zou, L.; Zhang, Y.; Liang, H. HIF-1α Inhibition Reverses Multidrug Resistance in Colon Cancer Cells via Downregulation of MDR1/P-Glycoprotein. PLoS ONE 2014, 9, e98882. [Google Scholar] [CrossRef] [PubMed]
- Terzic, J.; Abu el Maaty, M.A.; Lutzing, R.; Vincent, A.; El Bizri, R.; Jung, M.; Keime, C.; Metzger, D. Hypoxia-inducible Factor 1A Inhibition Overcomes Castration Resistance of Prostate Tumors. EMBO Mol. Med. 2023, 15, e17209. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Cao, H.-X.; He, Y.-W.; Ouyang, J.-J.; Lv, M.; Li, Y.-X.; Lu, Y.-D. Increased Keratin 80 Expression Predicts Poor Prognosis and Promotes Oxaliplatin Resistance in Gastric Cancer. World J. Gastroenterol. 2025, 31, 103991. [Google Scholar] [CrossRef]
- Yang, Q.-L.; Xie, Y.; Qiao, K.; Lim, J.Y.S.; Wu, S. Modern Biology of Extrachromosomal DNA: A Decade-Long Voyage of Discovery. Cell Res. 2025, 35, 11–22. [Google Scholar] [CrossRef]
- Bailey, C.; Pich, O.; Thol, K.; Watkins, T.B.K.; Luebeck, J.; Rowan, A.; Stavrou, G.; Weiser, N.E.; Dameracharla, B.; Bentham, R.; et al. Origins and Impact of Extrachromosomal DNA. Nature 2024, 635, 193–200. [Google Scholar] [CrossRef]
- Pal Choudhuri, S.; Girard, L.; Lim, J.Y.S.; Wise, J.F.; Freitas, B.; Yang, D.; Wong, E.; Hamilton, S.; Chien, V.D.; Kim, Y.J.; et al. Acquired Cross-Resistance in Small Cell Lung Cancer Due to Extrachromosomal DNA Amplification of MYC Paralogs. Cancer Discov. 2024, 14, 804–827. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Li, H.; Li, Q.; Li, Y.; Wu, H.; Ge, Y.; Zhu, X.; Zheng, Z.; Sun, Z. Characterization of Extrachromosomal Circular DNA in Primary and Cisplatin-Resistant High-Grade Serous Ovarian Cancer. Genes 2025, 16, 517. https://doi.org/10.3390/genes16050517
Wang Y, Li H, Li Q, Li Y, Wu H, Ge Y, Zhu X, Zheng Z, Sun Z. Characterization of Extrachromosomal Circular DNA in Primary and Cisplatin-Resistant High-Grade Serous Ovarian Cancer. Genes. 2025; 16(5):517. https://doi.org/10.3390/genes16050517
Chicago/Turabian StyleWang, Youya, He Li, Qinglan Li, Yi Li, Hao Wu, Yan Ge, Xingnuo Zhu, Zhiguo Zheng, and Zhongsheng Sun. 2025. "Characterization of Extrachromosomal Circular DNA in Primary and Cisplatin-Resistant High-Grade Serous Ovarian Cancer" Genes 16, no. 5: 517. https://doi.org/10.3390/genes16050517
APA StyleWang, Y., Li, H., Li, Q., Li, Y., Wu, H., Ge, Y., Zhu, X., Zheng, Z., & Sun, Z. (2025). Characterization of Extrachromosomal Circular DNA in Primary and Cisplatin-Resistant High-Grade Serous Ovarian Cancer. Genes, 16(5), 517. https://doi.org/10.3390/genes16050517