
Updated Gene Therapy for Renal Inborn Errors of Metabolism

 ,
,
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Epidemiology
3.2. Disorders of Amino Acid Metabolism
3.2.1. Methylmalonic Aciduria and Homocystinuria
3.2.2. Adenine Phosphoribosyltransferase Deficiency
3.2.3. Lysinuric Protein Intolerance
3.2.4. Argininemia
3.3. Disorders of Carbohydrate Metabolism
3.3.1. Glycogen Storage Diseases (Ia and Ib)
3.3.2. Fructose-1,6-Bisphophate Deficiency
3.4. Disorders of Energy Production
3.4.1. Coenzyme Q Deficiency
3.4.2. GRACILE Syndrome
3.4.3. Mitochondrial DNA Depletion Syndrome
3.5. Disorders of Organelle Function
3.5.1. Primary Hyperoxaluria
3.5.2. Lowe Oculocerebrorenal Syndrome
3.5.3. Fabry Disease
3.5.4. Congenital Adrenal Hyperplasia
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| IEM | inborn error of metabolism |
| CKD | chronic kidney disease |
| DDIEM | Drug Database for Inborn Errors of Metabolism |
| MMADHC | methylmalonic aciduria and homocystinuria |
| APRT | adenine phosphoribosyltransferase |
| GFR | glomerular filtration rate |
| UTI | urinary tract infection |
| AAV | adeno-associated virus |
| CAH | congenital adrenal hyperplasia |
| GRACILE | growth retardation, aminoaciduria, cholestasis, iron overload, lactic acidosis, early death |
| GSD | glycogen storage disease |
| HUS | hemolytic uremic syndrome |
References
- Ramoser, G.; Caferri, F.; Radlinger, B.; Brunner-Krainz, M.; Herbst, S.; Huemer, M.; Hufgard-Leitner, M.; Kircher, S.G.; Konstantopoulou, V.; Löscher, W.; et al. 100 years of inherited metabolic disorders in Austria—A national registry of minimal birth prevalence, diagnosis, and clinical outcome of inborn errors of metabolism in Austria between 1921 and 2021. J. Inherit. Metab. Dis. 2022, 45, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Bernardes, T.P.; Foresto, R.D.; Kirsztajn, G.M. Fabry disease: Genetics, pathology, and treatment. Rev. Assoc. Med. Bras. 2020, 66, s10–s16. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, S.; Green, A.; Preece, M.A.; Burton, H. The incidence of inherited metabolic disorders in the West Midlands, UK. Arch. Dis. Child. 2006, 91, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, U. Inborn Errors of Metabolism-Approach to Diagnosis and Management in Neonates. Indian J. Pediatr. 2021, 88, 679–689. [Google Scholar] [CrossRef]
- Ginn, S.L.; Mandwie, M.; Alexander, I.E.; Edelstein, M.; Abedi, M.R. Gene therapy clinical trials worldwide to 2023-an update. J. Gene Med. 2024, 26, e3721. [Google Scholar] [CrossRef]
- Jeanmonod, R.; Asuka, E.; Jeanmonod, D. Inborn Errors of Metabolism. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Deodato, F.; Boenzi, S.; Rizzo, C.; Abeni, D.; Caviglia, S.; Picca, S.; Bartuli, A.; Dionisi-Vici, C. Inborn errors of metabolism: An update on epidemiology and on neonatal-onset hyperammonemia. Acta Paediatr. Suppl. 2004, 93, 18–21. [Google Scholar] [CrossRef]
- Aliu, E.; Kanungo, S.; Arnold, G.L. Amino acid disorders. Ann. Transl. Med. 2018, 6, 471. [Google Scholar] [CrossRef]
- Arhip, L.; Brox-Torrecilla, N.; Romero, I.; Motilla, M.; Serrano-Moreno, C.; Miguélez, M.; Cuerda, C. Late-onset methylmalonic acidemia and homocysteinemia (cblC disease): Systematic review. Orphanet J. Rare Dis. 2024, 19, 20. [Google Scholar] [CrossRef]
- Sloan, J.L.; Carrillo, N.; Adams, D.; Venditti, C.P. Disorders of Intracellular Cobalamin Metabolism. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Chandler, R.J.; Venditti, C.P. Gene Therapy for Methylmalonic Acidemia: Past, Present, and Future. Hum. Gene Ther. 2019, 30, 1236–1244. [Google Scholar] [CrossRef]
- Chandler, R.J.; Di Pasquale, G.; Sloan, J.L.; McCoy, S.; Hubbard, B.T.; Kilts, T.M.; Manoli, I.; Chiorini, J.A.; Venditti, C.P. Systemic gene therapy for methylmalonic acidemia using the novel adeno-associated viral vector 44.9. Mol. Ther. Methods Clin. Dev. 2022, 27, 61–72. [Google Scholar] [CrossRef]
- May, F.J.; Head, P.E.; Venturoni, L.E.; Chandler, R.J.; Venditti, C.P. Central nervous system-targeted adeno-associated virus gene therapy in methylmalonic acidemia. Mol. Ther. Methods Clin. Dev. 2021, 21, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Bollée, G.; Dollinger, C.; Boutaud, L.; Guillemot, D.; Bensman, A.; Harambat, J.; Deteix, P.; Daudon, M.; Knebelmann, B.; Ceballos-Picot, I. Phenotype and genotype characterization of adenine phosphoribosyltransferase deficiency. J. Am. Soc. Nephrol. 2010, 21, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shingde, M.; Nankivell, B.J.; Tchan, M.C.; Bose, B.; Chapman, J.R.; Kable, K.; Kim, S.K.; Vucak-Dzumhur, M.; Wong, G.; et al. Adenine Phosphoribosyltransferase Deficiency: A Potentially Reversible Cause of CKD. Kidney Int. Rep. 2019, 4, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Rashid, I.; Verma, A.; Tiwari, P.; D’Cruz, S. Adenine phosphoribosyl transferase deficiency leads to renal allograft dysfunction in kidney transplant recipients: A systematic review. J. Bras. Nefrol. 2022, 44, 403–416. [Google Scholar] [CrossRef]
- Nunes, V.; Niinikoski, H. Lysinuric Protein Intolerance. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Mauhin, W.; Habarou, F.; Gobin, S.; Servais, A.; Brassier, A.; Grisel, C.; Roda, C.; Pinto, G.; Moshous, D.; Ghalim, F.; et al. Update on Lysinuric Protein Intolerance, a Multi-faceted Disease Retrospective cohort analysis from birth to adulthood. Orphanet J. Rare Dis. 2017, 12, 3. [Google Scholar] [CrossRef]
- Sebastio, G.; Sperandeo, M.P.; Andria, G. Lysinuric protein intolerance: Reviewing concepts on a multisystem disease. Am. J. Med. Genet. C Semin. Med. Genet. 2011, 157C, 54–62. [Google Scholar] [CrossRef]
- Bodoy, S.; Sotillo, F.; Espino-Guarch, M.; Sperandeo, M.P.; Ormazabal, A.; Zorzano, A.; Sebastio, G.; Artuch, R.; Palacín, M. Inducible Slc7a7 Knockout Mouse Model Recapitulates Lysinuric Protein Intolerance Disease. Int. J. Mol. Sci. 2019, 20, 5294. [Google Scholar] [CrossRef]
- Cohen, Y.H.; Bargal, R.; Zeigler, M.; Markus-Eidlitz, T.; Zuri, V.; Zeharia, A. Hyperargininemia: A Family with a Novel Mutation in an Unexpected Site. JIMD Rep. 2011, 5, 83–88. [Google Scholar] [CrossRef]
- Scaglia, F.; Lee, B. Clinical, biochemical, and molecular spectrum of hyperargininemia due to arginase I deficiency. Am. J. Med. Genet. C Semin. Med. Genet. 2006, 142C, 113–120. [Google Scholar] [CrossRef]
- Lee, E.K.; Hu, C.; Bhargava, R.; Ponnusamy, R.; Park, H.; Novicoff, S.; Rozengurt, N.; Marescau, B.; De Deyn, P.; Stout, D.; et al. AAV-based gene therapy prevents neuropathology and results in normal cognitive development in the hyperargininemic mouse. Gene Ther. 2013, 20, 785–796. [Google Scholar] [CrossRef]
- Hu, C.; Tai, D.S.; Park, H.; Cantero, G.; Cantero-Nieto, G.; Chan, E.; Yudkoff, M.; Cederbaum, S.D.; Lipshutz, G.S. Minimal ureagenesis is necessary for survival in the murine model of hyperargininemia treated by AAV-based gene therapy. Gene Ther. 2015, 22, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Stone, W.L.; John, T.A.; Anastasopoulou, C.; Basit, H.; Adil, A. Glycogen Storage Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Kishnani, P.S.; Austin, S.L.; Abdenur, J.E.; Arn, P.; Bali, D.S.; Boney, A.; Chung, W.K.; Dagli, A.I.; Dale, D.; Koeberl, D.; et al. Diagnosis and management of glycogen storage disease type I: A practice guideline of the American College of Medical Genetics and Genomics. Genet. Med. 2014, 16, e1. [Google Scholar] [CrossRef] [PubMed]
- Grinshpun, A.; Condiotti, R.; Waddington, S.N.; Peer, M.; Zeig, E.; Peretz, S.; Simerzin, A.; Chou, J.; Pann, C.-J.; Giladi, H.; et al. Neonatal Gene Therapy of Glycogen Storage Disease Type Ia Using a Feline Immunodeficiency Virus–based Vector. Mol. Ther. 2010, 18, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.Y.; Zingone, A.; Pan, C.-J. Adenovirus-mediated gene therapy in a mouse model of glycogen storage disease type 1a. Eur. J. Pediatr. 2002, 161, S56–S61. [Google Scholar] [CrossRef]
- Koeberl, D.D.; Sun, B.; Franco, L.; Bird, A.; Faulkner, E.; Chen, Y. 1054. Development of Adeno-Associated Virus (AAV) Vectors for Gene Therapy in the Mouse Model for Glycogen Storage Disease Type Ia (GSD-Ia). Mol. Ther. 2004, 9, S404. [Google Scholar] [CrossRef]
- Weinstein, D.A.; Derks, T.G.; Rodriguez-Buritica, D.F.; Ahmad, A.; Couce, M.-L.; Mitchell, J.J.; Riba-Wolman, R.; Mount, M.; Sallago, J.B.; Ross, K.M.; et al. Safety and Efficacy of DTX401, an AAV8-Mediated Liver-Directed Gene Therapy, in Adults With Glycogen Storage Disease Type I a (GSDIa). J. Inherit. Metab. Dis. 2025, 48, e70014. [Google Scholar] [CrossRef]
- Kwon, J.H.; Lee, Y.M.; Cho, J.-H.; Kim, G.-Y.; Anduaga, J.; Starost, M.F.; Mansfield, B.C.; Chou, J.Y. Liver-directed gene therapy for murine glycogen storage disease type Ib. Hum. Mol. Genet. 2017, 26, 4395–4405. [Google Scholar] [CrossRef]
- Moey, L.H.; Abdul Azize, N.A.; Yakob, Y.; Leong, H.Y.; Keng, W.T.; Chen, B.C.; Ngu, L.H. Fructose-1,6-bisphosphatase deficiency as a cause of recurrent hypoglycemia and metabolic acidosis: Clinical and molecular findings in Malaysian patients. Pediatr. Neonatol. 2018, 59, 397–403. [Google Scholar] [CrossRef]
- Bijarnia-Mahay, S.; Bhatia, S.; Arora, V. Fructose-1,6-Bisphosphatase Deficiency. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Mantle, D.; Millichap, L.; Castro-Marrero, J.; Hargreaves, I.P. Primary Coenzyme Q10 Deficiency: An Update. Antioxidants 2023, 12, 1652. [Google Scholar] [CrossRef]
- Salviati, L.; Trevisson, E.; Agosto, C.; Doimo, M.; Navas, P. Primary Coenzyme Q10 Deficiency Overview. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Xie, J.; Jiang, J.; Guo, Q. Primary Coenzyme Q10 Deficiency-7 and Pathogenic COQ4 Variants: Clinical Presentation, Biochemical Analyses, and Treatment. Front. Genet. 2021, 12, 776807. [Google Scholar] [CrossRef]
- Barriocanal-Casado, E.; Cueto-Ureña, C.; Benabdellah, K.; Gutiérrez-Guerrero, A.; Cobo, M.; Hidalgo-Gutiérrez, A.; Rodríguez-Sevilla, J.J.; Martín, F.; López, L.C. Gene Therapy Corrects Mitochondrial Dysfunction in Hematopoietic Progenitor Cells and Fibroblasts from Coq9R239X Mice. PLoS ONE 2016, 11, e0158344. [Google Scholar] [CrossRef] [PubMed]
- Fellman, V. GRACILE syndrome—A severe neonatal mitochondrial disorder. Duodecim 2012, 128, 1560–1567. [Google Scholar] [PubMed]
- Kasapkara, Ç.S.; Tümer, L.; Ezgü, F.S.; Küçükçongar, A.; Hasanoğlu, A. BCS1L gene mutation causing GRACILE syndrome: Case report. Ren. Fail. 2014, 36, 953–954. [Google Scholar] [CrossRef]
- Guo, W.; Shao, Y.; Lang, Y.; Wang, H.; Lin, Y.; Liu, X.; Zhang, R.; Shao, L. Identification of two novel variants of BCS1L gene in a patient with classical GRACILE syndrome. Nephrology 2022, 27, 810–814. [Google Scholar] [CrossRef]
- Lynn, A.M.; King, R.I.; Mackay, R.J.; Florkowski, C.M.; Wilson, C.J. BCS1L gene mutation presenting with GRACILE-like syndrome and complex III deficiency. Ann. Clin. Biochem. 2012, 49, 201–203. [Google Scholar] [CrossRef]
- Wang, Y.; Hu, L.-F.; Zhou, T.-J.; Qi, L.-Y.; Xing, L.; Lee, J.; Wang, F.-Z.; Oh, Y.-K.; Jiang, H.-L. Gene therapy strategies for rare monogenic disorders with nuclear or mitochondrial gene mutations. Biomaterials 2021, 277, 121108. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial DNA Depletion Syndromes: Review and Updates of Genetic Basis, Manifestations, and Therapeutic Options. Neurotherapeutics 2013, 10, 186–198. [Google Scholar] [CrossRef]
- Morava, E.; Steuerwald, U.; Carrozzo, R.; Kluijtmans, L.A.J.; Joensen, F.; Santer, R.; Dionisi-Vici, C.; Wevers, R.A. Dystonia and deafness due to SUCLA2 defect; Clinical course and biochemical markers in 16 children. Mitochondrion 2009, 9, 438–442. [Google Scholar] [CrossRef]
- Sas, D.J.; Enders, F.T.; Mehta, R.A.; Tang, X.; Zhao, F.; Seide, B.M.; Milliner, D.S.; Lieske, J.C. Clinical features of genetically confirmed patients with primary hyperoxaluria identified by clinical indication versus familial screening. Kidney Int. 2020, 97, 786–792. [Google Scholar] [CrossRef]
- Wood, K.D.; Freeman, B.L.; Killian, M.E.; Lai, W.S.; Assimos, D.; Knight, J.; Fargue, S. Effect of alanine supplementation on oxalate synthesis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 165981. [Google Scholar] [CrossRef]
- Salido, E.; Rodriguez-Pena, M.; Santana, A.; Beattie, S.G.; Petry, H.; Torres, A. Phenotypic correction of a mouse model for primary hyperoxaluria with adeno-associated virus gene transfer. Mol. Ther. 2011, 19, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Dutta, C.; Avitahl-Curtis, N.; Pursell, N.; Larsson Cohen, M.; Holmes, B.; Diwanji, R.; Zhou, W.; Apponi, L.; Koser, M.; Ying, B.; et al. Inhibition of Glycolate Oxidase With Dicer-substrate siRNA Reduces Calcium Oxalate Deposition in a Mouse Model of Primary Hyperoxaluria Type 1. Mol. Ther. 2016, 24, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Sena, C.; Iannello, G.; Skowronski, A.A.; Dannheim, K.; Cheung, L.; Agrawal, P.B.; Hirschhorn, J.N.; Zeitler, P.; LeDuc, C.A.; Stratigopoulos, G.; et al. Endocrine and behavioural features of Lowe syndrome and their potential molecular mechanisms. J. Med. Genet. 2022, 59, 1171–1178. [Google Scholar] [CrossRef]
- Bökenkamp, A.; Ludwig, M. The oculocerebrorenal syndrome of Lowe: An update. Pediatr. Nephrol. 2016, 31, 2201–2212. [Google Scholar] [CrossRef]
- Chen, S.; Lo, C.-H.; Liu, Z.; Wang, Q.; Ning, K.; Li, T.; Sun, Y. Base editing correction of OCRL in Lowe syndrome: ABE-mediated functional rescue in patient-derived fibroblasts. Hum. Mol. Genet. 2024, 33, 1142–1151. [Google Scholar] [CrossRef]
- Khan, A.; Barber, D.L.; Huang, J.; Rupar, C.A.; Rip, J.W.; Auray-Blais, C.; Boutin, M.; O’Hoski, P.; Gargulak, K.; McKillop, W.M.; et al. Lentivirus-mediated gene therapy for Fabry disease. Nat. Commun. 2021, 12, 1178. [Google Scholar] [CrossRef]
- Hopkin, R.J.; Bissler, J.; Banikazemi, M.; Clarke, L.; Eng, C.M.; Germain, D.P.; Lemay, R.; Tylki-Szymanska, A.; Wilcox, W.R. Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. Pediatr. Res. 2008, 64, 550–555. [Google Scholar] [CrossRef]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef]
- Wan, Z.; Wang, W.; Zheng, S.; Han, R.; Xie, X.; Zhao, Y.; Wang, W.; Sun, S.; Ye, L. Nonclassic Adrenal Hyperplasia (NCAH) due to 21-hydroxylase deficiency: A cohort of 78 patients. J. Steroid Biochem. Mol. Biol. 2023, 225, 106192. [Google Scholar] [CrossRef]
- Naiki, Y.; Miyado, M.; Horikawa, R.; Katsumata, N.; Onodera, M.; Pang, S.; Ogata, T.; Fukami, M. Extra-adrenal induction of Cyp21a1 ameliorates systemic steroid metabolism in a mouse model of congenital adrenal hyperplasia. Endocr. J. 2016, 63, 897–904. [Google Scholar] [CrossRef]
- Markmann, S.; De, B.P.; Reid, J.; Jose, C.L.; Rosenberg, J.B.; Leopold, P.L.; Kaminsky, S.M.; Sondhi, D.; Pagovich, O.; Crystal, R.G. Biology of the Adrenal Gland Cortex Obviates Effective Use of Adeno-Associated Virus Vectors to Treat Hereditary Adrenal Disorders. Hum. Gene Ther. 2018, 29, 403–412. [Google Scholar] [CrossRef]
- Schumann, A.; Schultheiss, U.T.; Ferreira, C.R.; Blau, N. Clinical and biochemical footprints of inherited metabolic diseases. XIV. Metabolic kidney diseases. Mol. Genet. Metab. 2023, 140, 107683. [Google Scholar] [CrossRef] [PubMed]
- Parasyri, M.; Brandström, P.; Uusimaa, J.; Ostergaard, E.; Hikmat, O.; Isohanni, P.; Naess, K.; de Coo, I.F.M.; Nascimento Osorio, A.; Nuutinen, M.; et al. Renal Phenotype in Mitochondrial Diseases: A Multicenter Study. Kidney Dis. 2022, 8, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Morath, M.A.; Hörster, F.; Sauer, S.W. Renal dysfunction in methylmalonic acidurias: Review for the pediatric nephrologist. Pediatr. Nephrol. 2013, 28, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Edvardsson, V.O.; Goldfarb, D.S.; Lieske, J.C.; Beara-Lasic, L.; Anglani, F.; Milliner, D.S.; Palsson, R. Hereditary causes of kidney stones and chronic kidney disease. Pediatr. Nephrol. 2013, 28, 1923–1942. [Google Scholar] [CrossRef]
- Runolfsdottir, H.L.; Palsson, R.; Agustsdottir, I.M.; Indridason, O.S.; Edvardsson, V.O. Long-term renal outcomes of APRT deficiency presenting in childhood. Pediatr. Nephrol. 2019, 34, 435–442. [Google Scholar] [CrossRef]
- Kubihal, S.; Goyal, A.; Singla, R.; Khadgawat, R. Urolithiasis due to Hereditary Xanthinuria Type II: A Long-term Follow-up report. Indian Pediatr. 2020, 57, 468–469. [Google Scholar] [CrossRef]
- Estève, E.; Krug, P.; Hummel, A.; Arnoux, J.-B.; Boyer, O.; Brassier, A.; de Lonlay, P.; Vuiblet, V.; Gobin, S.; Salomon, R.; et al. Renal involvement in lysinuric protein intolerance: Contribution of pathology to assessment of heterogeneity of renal lesions. Hum. Pathol. 2017, 62, 160–169. [Google Scholar] [CrossRef]
- Lugin, M.L.; Lee, R.T.; Kwon, Y.J. Synthetically Engineered Adeno-Associated Virus for Efficient, Safe, and Versatile Gene Therapy Applications. ACS Nano 2020, 14, 14262–14283. [Google Scholar] [CrossRef]
- Zaman, H.; Khan, A.; Khan, K.; Toheed, S.; Abdullah, M.; Zeeshan, H.M.; Hameed, A.; Umar, M.; Shahid, M.; Malik, K.; et al. Adeno-Associated Virus-Mediated Gene Therapy. Crit. Rev. Eukaryot. Gene Expr. 2023, 33, 87–100. [Google Scholar] [CrossRef]
- Wang, J.-H.; Gessler, D.J.; Zhan, W.; Gallagher, T.L.; Gao, G. Adeno-associated virus as a delivery vector for gene therapy of human diseases. Signal Transduct. Target. Ther. 2024, 9, 78. [Google Scholar] [CrossRef] [PubMed]
- Zwi-Dantsis, L.; Mohamed, S.; Massaro, G.; Moeendarbary, E. Adeno-Associated Virus Vectors: Principles, Practices, and Prospects in Gene Therapy. Viruses 2025, 17, 239. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.A.; Rodino-Klapac, L.R.; Goodspeed, K.; Gray, S.J.; Kay, C.N.; Boye, S.L.; Boye, S.E.; George, L.A.; Salabarria, S.; et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol. Ther. 2021, 29, 464–488. [Google Scholar] [CrossRef] [PubMed]
- Warrington, K.H.; Herzog, R.W. Treatment of human disease by adeno-associated viral gene transfer. Hum. Genet. 2006, 119, 571–603. [Google Scholar] [CrossRef]
- Pontoizeau, C.; Simon-Sola, M.; Gaborit, C.; Nguyen, V.; Rotaru, I.; Tual, N.; Colella, P.; Girard, M.; Biferi, M.-G.; Arnoux, J.-B.; et al. Neonatal gene therapy achieves sustained disease rescue of maple syrup urine disease in mice. Nat. Commun. 2022, 13, 3278. [Google Scholar] [CrossRef]
- Celik, B.; Rintz, E.; Sansanwal, N.; Khan, S.; Bigger, B.; Tomatsu, S. Lentiviral Vector-Mediated Ex Vivo Hematopoietic Stem Cell Gene Therapy for Mucopolysaccharidosis IVA Murine Model. Hum. Gene Ther. 2024, 35, 917–937. [Google Scholar] [CrossRef]
- Bondue, T.; van den Heuvel, L.; Levtchenko, E.; Brock, R. The potential of RNA-based therapy for kidney diseases. Pediatr. Nephrol. 2023, 38, 327–344. [Google Scholar] [CrossRef]
- Peek, J.L.; Wilson, M.H. Cell and gene therapy for kidney disease. Nat. Rev. Nephrol. 2023, 19, 451–462. [Google Scholar] [CrossRef]
- Baruteau, J.; Brunetti-Pierri, N.; Gissen, P. Liver-directed gene therapy for inherited metabolic diseases. J. Inherit. Metab. Dis. 2024, 47, 9–21. [Google Scholar] [CrossRef]
- Ginocchio, V.M.; Ferla, R.; Auricchio, A.; Brunetti-Pierri, N. Current Status on Clinical Development of Adeno-Associated Virus-Mediated Liver-Directed Gene Therapy for Inborn Errors of Metabolism. Hum. Gene Ther. 2019, 30, 1204–1210. [Google Scholar] [CrossRef]
- Chiesa, R.; Bernardo, M.E. Haematopoietic stem cell gene therapy in inborn errors of metabolism. Br. J. Haematol. 2022, 198, 227–243. [Google Scholar] [CrossRef]

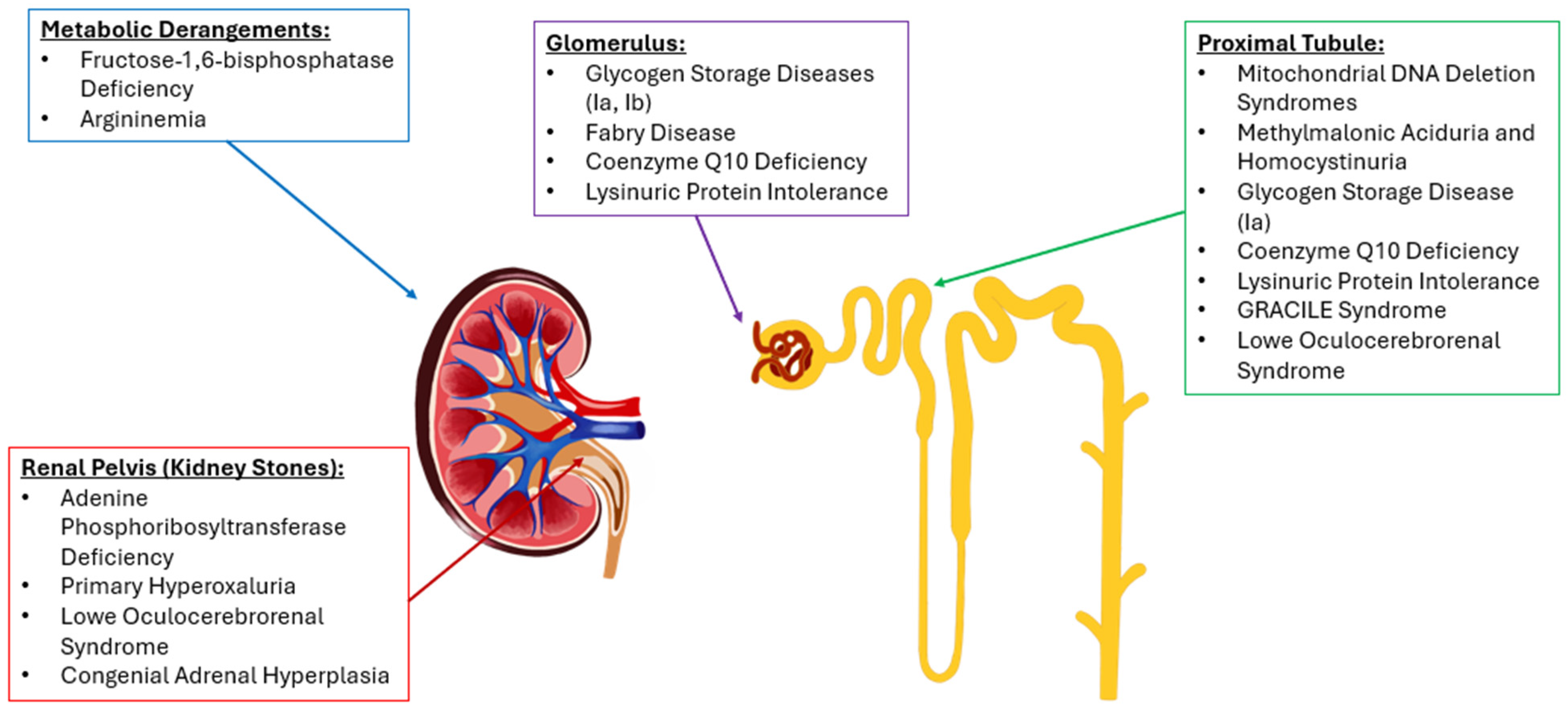

{kind=link}
{kind=link}
| Disease Name | Treatment/Gene Therapy | Phenotype Corrected |
|---|---|---|
| Methylmalonic Aciduria and Homocystinuria (Types C and D) | C: Leucovorin + glycine betaine + vitamin B12 D: Vit B12 + glycine betaine + folic acid, protein restriction, carglumic acid | C: Impaired renal function, decreased GFR, elevated total plasma homocysteine, high urine MMA level D: Elevated plasma non-protein-bound homocysteine (hyperhomocystinemia), hypomethioninemia, hyperammonemia |
| Adenine Phosphoribosyltransferase Deficiency | Allopurinol/Febuxostat | Dysuria, CKD, recurrent UTIs, urolithiasis, hematuria, crystalline nephropathy, elevated urinary 2,8-dihydroxyadenine (DHA) excretion |
| Lysinuric Protein Intolerance | L-citrulline, Ciclosporin + Amcinonide, phenylbutyric acid, benzoic acid, L-lysine, benazepril | Hyperammonemia, hemophagocytic lymphohistiocytosis, kidney failure, proteinuria, hypertension, orotic aciduria |
| Argininemia | Benzoic acid, phenylbutyric acid Gene Therapy: Aav-based gene therapy, Aeb1102(pegzilarginase) Co-argi-peg Modified Human Arginase I | Neurotoxicity, hyperammonemia |
| Glycogen Storage Diseases (Ia and Ib) | I: Alglucosidase Alfa II: High-fat and high-protein diet Gene Therapy: Ia: Dtx401, Fiv-haat-g6pase, Ad-mg6pase, Aav-cg6pgh, Adhd-g6pase Ib: Raav-gpe-g6pt | Hypertrophic cardiomyopathy, mortality II: Elevated CK, weakness 1a, Ib: enlarged kidney, growth failure, lactic acidemia, hyperuricemia |
| Fructose-1,6-bisphosphatase Deficiency | D-glucose | Hypoglycemia |
| Coenzyme Q10 Deficiency | Ubidecarenone | Hyperlactatemia, exercise intolerance |
| GRACILE Syndrome | Sodium bicarbonate | Lactic acidosis |
| Mitochondrial DNA Depletion Syndromes (4A, 5, 7) | 4A: Magnesium, Vatiquinone 5: Vitamin B1 + B2 7: Levocarnitine, glutathione | 4A: Seizures 5: Oxidative stress 7: Abnormal circulating creatinine level, oxidative stress |
| Fabry Disease | Pegunigalsidase Alfa + Agalsidase β, Lucerastat, Migalastat | Abnormal kidney function, high Gb3 accumulation, high mean number of GL-3 inclusions, kidney interstitial capillary |
| Primary Hyperoxaluria | Pyridoxine + calcium oxalate crystallization inhibitors, pyridoxine, magnesium hydroxide, sodium citrate, potassium citrate, oxalobacter Formigenes Gene Therapy: Aav8-agxt and Aav5-agxt, helper-dependent adenoviral vectors for liver-directed gene therapy, deno-associated virus carrying one copy of Agxt Cdna, polymer-conjugated Agt | Nephrocalcinosis, urolithiasis, abnormal kidney function, high urine oxalate level |
| Lowe Oculocerebrorenal Syndrome | Sodium bicarbonate, potassium bicarbonate, potassium citrate, Quinethazone, Levocarnitine Gene Therapy: ABE8e-NG-based correction | Proximal tubular dysfunction, high calcium excretion, hypokalemia, metabolic acidosis, secondary carnitine deficiency |
| Congenital Adrenal Hyperplasia (21-hydroxylase Deficiency) | Hydrocortisone, Dexamethasone Gene Therapy: Aavrh.10-21oh-ha, Adeno-associated viral vector containing Cyp21a1 | Hypertension, abnormal circulating renin, aldosterone |
| Disease Name | Gene Affected | Enzyme Deficient |
|---|---|---|
| Methylmalonic aciduria and Homocystinuria (Types C and D) | MMADHC | Methylmalonic aciduria and homocystinuria type C/D protein |
| Adenine Phosphoribosyltransferase Deficiency | APRT | Adenine phosphoribosyltransferase |
| Lysinuric Protein Intolerance | SLC7A7 | Y+L Amino Acid Transporter 1 |
| Argininemia | ARG1 | Arginase |
| Glycogen Storage Diseases (Ia and Ib) | Ia: G6PC Ib: SLC37A4 | I: 7-dehydrocholesterol reductase Ib: Glucose-6-phosphate exchanger SLC37A4 (glucose 6-phosphate translocase) |
| Fructose-1,6-bisphosphatase Deficiency | FBP1 | Fructose-1,6-bisphosphatase 1 |
| Coenzyme Q10 Deficiency | CABC1, ADCK3 | Atypical Kinase COQ8A (mitochondrial) |
| GRACILE Syndrome | BCS1L | Ubiquinol–cytochrome c reductase complex chaperone |
| Mitochondrial DNA Depletion Syndromes | 4A: POLG 5: SUCLA2 7: TWNK | 4A: DNA polymerase subunit γ-1 5: Succinyl-CoA synthetase (β subunit) 7: Twinkle mtDNA helicase |
| Fabry Disease | GLA | α-Galactosidase A |
| Primary Hyperoxaluria | AGXT | Serine–pyruvate aminotransferase |
| Lowe Oculocerebrorenal Syndrome | OCRL | Inositol polyphosphate 5-phosphatase OCRL-1 |
| Congenital Adrenal Hyperplasia (21-hydroxylase Deficiency) | CYP17A1 CYP21A1 | 17-α hydroxylase Frataxin |
| Disease Name | Age of Onset | Diagnostic Tests |
|---|---|---|
| Methylmalonic aciduria and Homocystinuria (Type C and D) | C: Birth D: <2 years | Plasma Analysis: Elevated methylmalonic acid or homocysteine Genetic Testing: Mutations in MMADHC or MMACHC |
| Adenine Phosphoribosyltransferase Deficiency | 6 months–72 years (mean = 36.3 years) | Microscopy: 2,8-dihydroxyadenine (DHA) crystals in urine Mass spectrometry: UPLC-MS/MS assay Genetic Testing: Mutations of APRT gene |
| Lysinuric Protein Intolerance | 6–12 months (once weaned from breast milk) | Urinalysis: Elevated lysine, ornithine, and arginine in urine Genetic Testing: Mutations in SLC7A7 gene |
| Argininemia | 3 years | Newborn Screen (NBS) Plasma Analysis: Elevated arginine or ammonia Genetic Testing: Mutations in ARG1 gene |
| Glycogen Storage Diseases (Ia and Ib) | 3–6 months | Enzyme Assay: Decreased G6Pase (Ia) or G6P-translocase (Ib) activity Genetic Testing: Mutations in G6PC (Ia) or SLC37A4 (Ib) genes |
| Fructose-1,6-bisphosphatase Deficiency | <1 year (often within first week) | Genetic Testing: Mutations in FBP1 gene |
| Coenzyme Q10 Deficiency | Birth–70 years | Muscle Biopsy: Reduced CoQ10 activity Genetic Testing: Mutations in COQ genes (PDSS1, PDSS2, COQ2, COQ4, COQ6, ADCK3, ADCK4, COQ9) |
| GRACILE Syndrome | Birth | Genetic Testing: Mutations in BCS1L gene |
| Mitochondrial DNA Depletion Syndromes (4A, 5, 7) | Birth–2 years | PCR: mtDNA deletions Genetic Testing: Mutations in POLG (4a), SUCLA2 (5), and TWNK (7) genes |
| Fabry Disease | Males: Average = 6 years Females: Average = 9 years | Enzyme Assay: Decreased α-galactosidase A Genetic Testing: Mutations in GLA gene |
| Primary Hyperoxaluria | Average = 3–4 years | Urinalysis: Elevated oxalate, glycolate, or glycerate Genetic Testing: Mutations in AGXT |
| Lowe Oculocerebrorenal Syndrome | Birth | Genetic Testing: Mutations in OCRL gene |
| Congenital Adrenal Hyperplasia (21-hydroxylase Deficiency) | Classic: Birth–12 months Nonclassical Males: 11 years Nonclassical Females: 13 years | Plasma Analysis: Elevated 17-hydroxyprogesterone and 21-deoxycortisol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hergenrother, S.; Husein, M.; Thompson, C.; Kalina, E.; Raina, R. Updated Gene Therapy for Renal Inborn Errors of Metabolism. Genes 2025, 16, 516. https://doi.org/10.3390/genes16050516
Hergenrother S, Husein M, Thompson C, Kalina E, Raina R. Updated Gene Therapy for Renal Inborn Errors of Metabolism. Genes. 2025; 16(5):516. https://doi.org/10.3390/genes16050516
Chicago/Turabian StyleHergenrother, Sean, Mustafa Husein, Cole Thompson, Ethan Kalina, and Rupesh Raina. 2025. "Updated Gene Therapy for Renal Inborn Errors of Metabolism" Genes 16, no. 5: 516. https://doi.org/10.3390/genes16050516
APA StyleHergenrother, S., Husein, M., Thompson, C., Kalina, E., & Raina, R. (2025). Updated Gene Therapy for Renal Inborn Errors of Metabolism. Genes, 16(5), 516. https://doi.org/10.3390/genes16050516