

Epigenetic Regulation of Human Vascular Calcification
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
Abstract
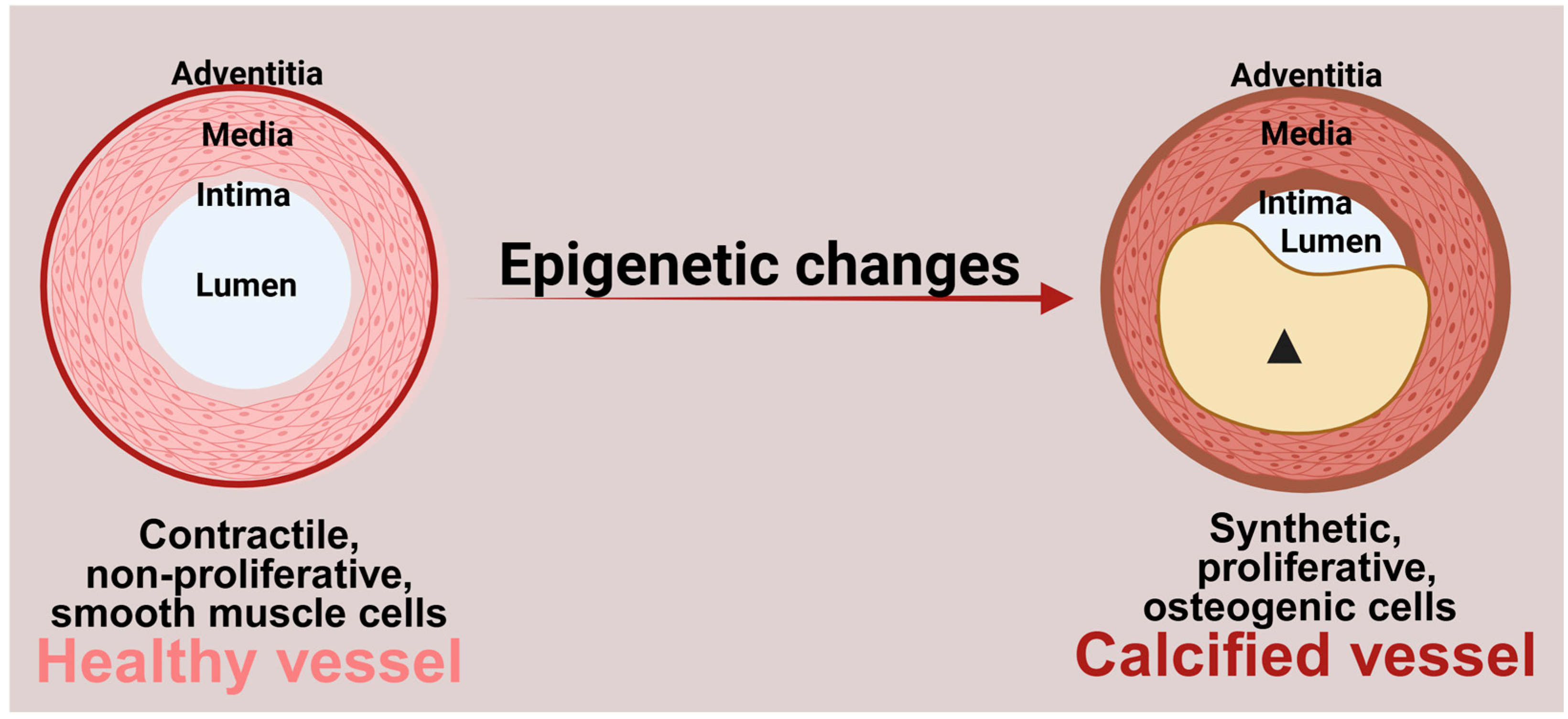
1. Introduction
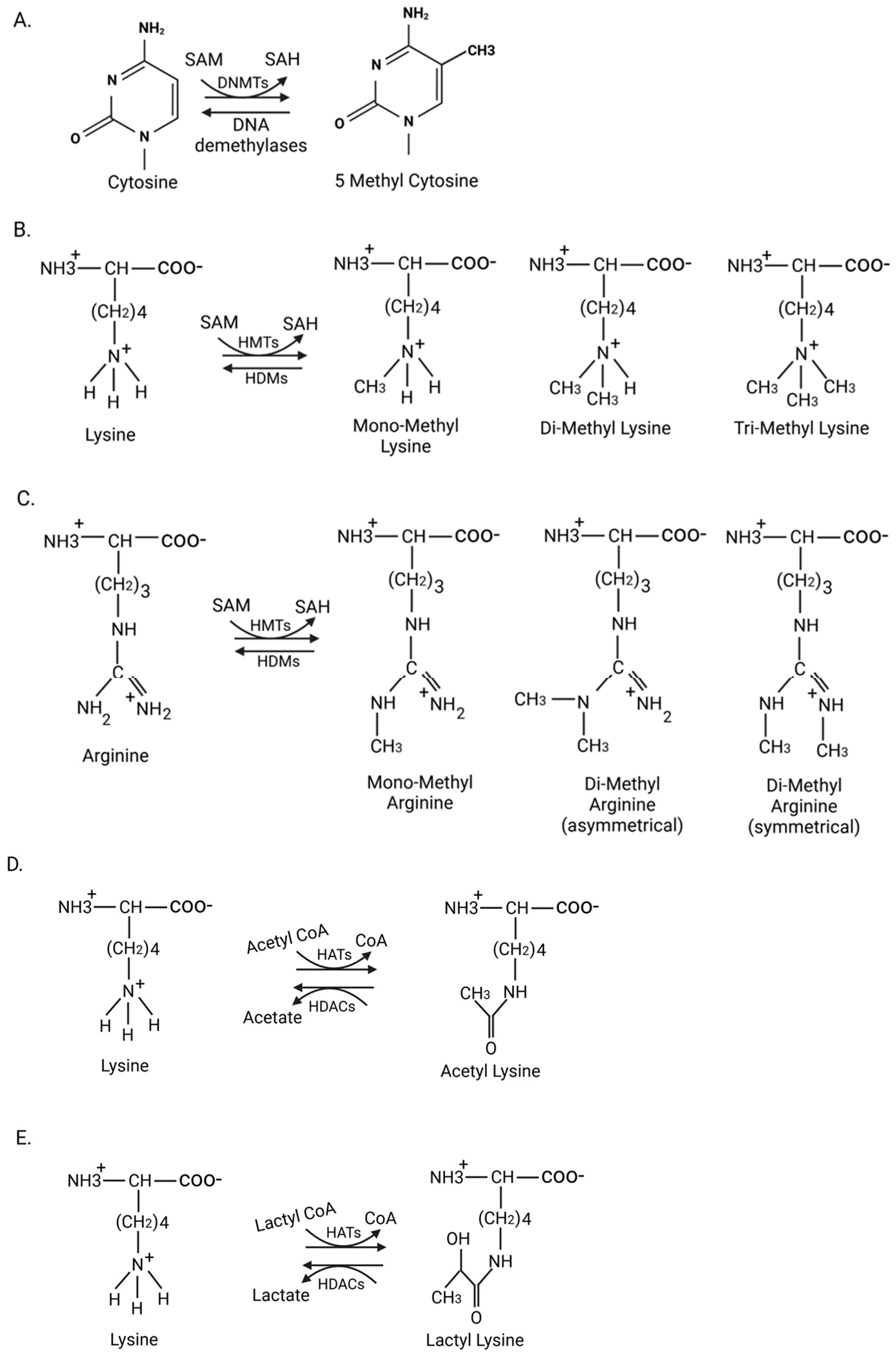
2. DNA Methylation
3. Histone Modifications
3.1. Histone Methylation
3.2. Histone Acetylation
3.3. Histone Lactylation
4. Non-Coding RNAs
4.1. Long Non-Coding RNAs
4.2. Small Non-Coding RNAs
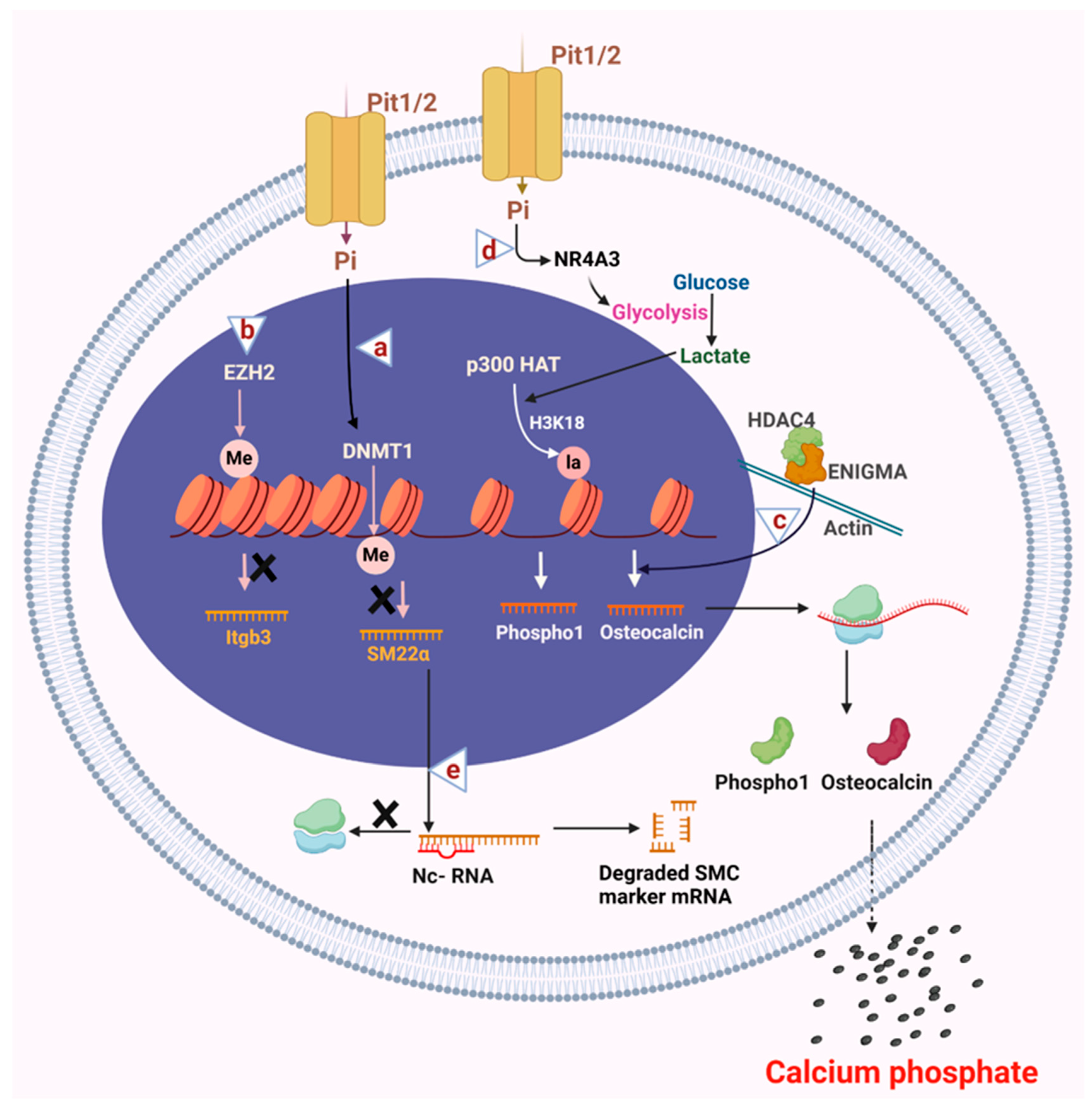
5. Targeting Epigenetic Regulators to Treat Vascular Calcification
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AGE | Aged garlic extract |
| αSMA | Alpha-smooth muscle actin |
| ALP | Alkaline phosphatase |
| ANCR | Angelman syndrome chromosome region |
| 5aza-2dc | 5-aza-2′-deoxycytidine |
| BMPs | Bone morphogenic proteins |
| CVDs | Cardiovascular diseases |
| DDR1 | Discoidin domain receptor 1 |
| DNMTs | DNA methyltransferases |
| ECM | Extracellular matrix |
| ERK | Extracellular signal-regulated kinase |
| EZH2 | Enhancer of zeste homolog 2 |
| FOXO | Forkhead box O |
| GAS5 | Growth arrest-specific 5 |
| HATs | Histone acetyltransferases |
| HDACs | Histone deacetylases |
| HMTs | Histone methyl transferases |
| IL-6 | Interleukin-6 |
| LINC | Linker of the nucleoskeleton and cytoskeleton complex |
| lncRNAs | Long non-coding RNAs |
| lincRNA-EPS | Long intergenic RNA-erythroid pro-survival |
| LRP6 | Lipoprotein receptor-related protein 6 |
| MALAT1 | Metastasis associated lung adenocarcinoma transcript 1 |
| MAPK | Mitogen-activated protein kinase |
| NF-κB | Nuclear factor kappa B |
| NR4A3 | Nuclear receptor subfamily 4 group A member |
| PRC2 | Polycomb repressive complex 2 |
| PTEN | Phosphate and tensin homolog |
| RUNX2 | Runt-related transcription factor 2 |
| sIL-6R | Soluble interleukin-6 receptor |
| SM22α | Smooth muscle protein 22-alpha |
| SNAI2 | Snail family transcriptional repressor 2 |
| SNHG1 | Small nucleolar RNA host gene 1 |
| STAT3 | Signal transducer and activator of transcription 3 |
| TLR3 | Toll like receptor 3 |
| TUG1 | Taurine upregulated 1 |
| VSMCs | Vascular smooth muscle cells |
| Wnt | Wingless integrated |
References
- Mocumbi, A.O. Cardiovascular Health Care in Low- and Middle-Income Countries. Circulation 2024, 149, 557–559. [Google Scholar] [CrossRef]
- Piché, M.E.; Tchernof, A.; Després, J.P. Obesity Phenotypes, Diabetes, and Cardiovascular Diseases. Circ. Res. 2020, 126, 1477–1500. [Google Scholar] [CrossRef]
- Joynt Maddox, K.E.; Elkind, M.S.V.; Aparicio, H.J.; Commodore-Mensah, Y.; de Ferranti, S.D.; Dowd, W.N.; Hernandez, A.F.; Khavjou, O.; Michos, E.D.; Palaniappan, L.; et al. Forecasting the Burden of Cardiovascular Disease and Stroke in the United States Through 2050-Prevalence of Risk Factors and Disease: A Presidential Advisory From the American Heart Association. Circulation 2024, 150, e65–e88. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Fan, J.; Watanabe, T. Atherosclerosis: Known and unknown. Pathol. Int. 2022, 72, 151–160. [Google Scholar] [CrossRef]
- Onnis, C.; Virmani, R.; Kawai, K.; Nardi, V.; Lerman, A.; Cademartiri, F.; Scicolone, R.; Boi, A.; Congiu, T.; Faa, G.; et al. Coronary Artery Calcification: Current Concepts and Clinical Implications. Circulation 2024, 149, 251–266. [Google Scholar] [CrossRef]
- Sutton, N.R.; Malhotra, R.; St Hilaire, C.; Aikawa, E.; Blumenthal, R.S.; Gackenbach, G.; Goyal, P.; Johnson, A.; Nigwekar, S.U.; Shanahan, C.M.; et al. Molecular Mechanisms of Vascular Health: Insights From Vascular Aging and Calcification. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 15–29. [Google Scholar] [CrossRef]
- Elmarasi, M.; Elmakaty, I.; Elsayed, B.; Elsayed, A.; Zein, J.A.; Boudaka, A.; Eid, A.H. Phenotypic switching of vascular smooth muscle cells in atherosclerosis, hypertension, and aortic dissection. J. Cell Physiol. 2024, 239, e31200. [Google Scholar] [CrossRef]
- Tyson, J.; Bundy, K.; Roach, C.; Douglas, H.; Ventura, V.; Segars, M.F.; Schwartz, O.; Simpson, C.L. Mechanisms of the Osteogenic Switch of Smooth Muscle Cells in Vascular Calcification: WNT Signaling, BMPs, Mechanotransduction, and EndMT. Bioengineering 2020, 7, 88. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Byon, C.H.; Yuan, K.; Chen, J.; Mao, X.; Heath, J.M.; Javed, A.; Zhang, K.; Anderson, P.G.; Chen, Y. Smooth muscle cell-specific runx2 deficiency inhibits vascular calcification. Circ. Res. 2012, 111, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Hruska, K.A.; Mathew, S.; Saab, G. Bone Morphogenetic Proteins in Vascular Calcification. Circ. Res. 2005, 97, 105–114. [Google Scholar] [CrossRef]
- Steitz, S.A.; Speer, M.Y.; Curinga, G.; Yang, H.Y.; Haynes, P.; Aebersold, R.; Schinke, T.; Karsenty, G.; Giachelli, C.M. Smooth muscle cell phenotypic transition associated with calcification: Upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ. Res. 2001, 89, 1147–1154. [Google Scholar] [CrossRef]
- Bundy, K.; Boone, J.; Simpson, C.L. Wnt Signaling in Vascular Calcification. Front. Cardiovasc. Med. 2021, 8, 708470. [Google Scholar] [CrossRef]
- Shimizu, T.; Tanaka, T.; Iso, T.; Doi, H.; Sato, H.; Kawai-Kowase, K.; Arai, M.; Kurabayashi, M. Notch Signaling Induces Osteogenic Differentiation and Mineralization of Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1104–1111. [Google Scholar] [CrossRef]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef]
- Patil, V.S.; Zhou, R.; Rana, T.M. Gene regulation by non-coding RNAs. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 16–32. [Google Scholar] [CrossRef]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Jin, B.; Robertson, K.D. DNA methyltransferases, DNA damage repair, and cancer. Adv. Exp. Med. Biol. 2013, 754, 3–29. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Mattei, A.L.; Bailly, N.; Meissner, A. DNA methylation: A historical perspective. Trends Genet. 2022, 38, 676–707. [Google Scholar] [CrossRef]
- Hutcheson, J.D.; Goettsch, C. Cardiovascular Calcification Heterogeneity in Chronic Kidney Disease. Circ. Res. 2023, 132, 993–1012. [Google Scholar] [CrossRef]
- Palit, S.; Kendrick, J. Vascular calcification in chronic kidney disease: Role of disordered mineral metabolism. Curr. Pharm. Des. 2014, 20, 5829–5833. [Google Scholar] [CrossRef]
- Dube, P.; DeRiso, A.; Patel, M.; Battepati, D.; Khatib-Shahidi, B.; Sharma, H.; Gupta, R.; Malhotra, D.; Dworkin, L.; Haller, S.; et al. Vascular Calcification in Chronic Kidney Disease: Diversity in the Vessel Wall. Biomedicines 2021, 9, 404. [Google Scholar] [CrossRef]
- Montes de Oca, A.; Madueño, J.A.; Martinez-Moreno, J.M.; Guerrero, F.; Muñoz-Castañeda, J.; Rodriguez-Ortiz, M.E.; Mendoza, F.J.; Almaden, Y.; Lopez, I.; Rodriguez, M.; et al. High-phosphate-induced calcification is related to SM22α promoter methylation in vascular smooth muscle cells. J. Bone Miner. Res. 2010, 25, 1996–2005. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, X.; Zhang, H.; Liu, T.; Zhang, H.; Teng, J.; Ji, J.; Ding, X. Indoxyl Sulfate Enhance the Hypermethylation of Klotho and Promote the Process of Vascular Calcification in Chronic Kidney Disease. Int. J. Biol. Sci. 2016, 12, 1236–1246. [Google Scholar] [CrossRef]
- Scheinbart, L.S.; Johnson, M.A.; Gross, L.A.; Edelstein, S.R.; Richardson, B.C. Procainamide inhibits DNA methyltransferase in a human T cell line. J. Rheumatol. 1991, 18, 530–534. [Google Scholar]
- Creusot, F.; Acs, G.; Christman, J.K. Inhibition of DNA methyltransferase and induction of Friend erythroleukemia cell differentiation by 5-azacytidine and 5-aza-2′-deoxycytidine. J. Biol. Chem. 1982, 257, 2041–2048. [Google Scholar] [CrossRef]
- Wang, J.C.; Bennett, M. Aging and Atherosclerosis. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef]
- Head, T.; Daunert, S.; Goldschmidt-Clermont, P.J. The Aging Risk and Atherosclerosis: A Fresh Look at Arterial Homeostasis. Front. Genet. 2017, 8, 216. [Google Scholar] [CrossRef]
- Wagenseil, J.E.; Mecham, R.P. Vascular extracellular matrix and arterial mechanics. Physiol. Rev. 2009, 89, 957–989. [Google Scholar] [CrossRef]
- Oh, Y.S.; Berkowitz, D.E.; Cohen, R.A.; Figueroa, C.A.; Harrison, D.G.; Humphrey, J.D.; Larson, D.F.; Leopold, J.A.; Mecham, R.P.; Ruiz-Opazo, N.; et al. A Special Report on the NHLBI Initiative to Study Cellular and Molecular Mechanisms of Arterial Stiffness and Its Association With Hypertension. Circ. Res. 2017, 121, 1216–1218. [Google Scholar] [CrossRef]
- Xie, S.A.; Zhang, T.; Wang, J.; Zhao, F.; Zhang, Y.P.; Yao, W.J.; Hur, S.S.; Yeh, Y.T.; Pang, W.; Zheng, L.S.; et al. Matrix stiffness determines the phenotype of vascular smooth muscle cell in vitro and in vivo: Role of DNA methyltransferase 1. Biomaterials 2018, 155, 203–216. [Google Scholar] [CrossRef]
- Di, X.; Gao, X.; Peng, L.; Ai, J.; Jin, X.; Qi, S.; Li, H.; Wang, K.; Luo, D. Cellular mechanotransduction in health and diseases: From molecular mechanism to therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 282. [Google Scholar] [CrossRef]
- Davis, M.J.; Earley, S.; Li, Y.S.; Chien, S. Vascular mechanotransduction. Physiol. Rev. 2023, 103, 1247–1421. [Google Scholar] [CrossRef]
- Wang, J.; Xie, S.A.; Li, N.; Zhang, T.; Yao, W.; Zhao, H.; Pang, W.; Han, L.; Liu, J.; Zhou, J. Matrix stiffness exacerbates the proinflammatory responses of vascular smooth muscle cell through the DDR1-DNMT1 mechanotransduction axis. Bioact. Mater. 2022, 17, 406–424. [Google Scholar] [CrossRef]
- Gross, D.S.; Chowdhary, S.; Anandhakumar, J.; Kainth, A.S. Chromatin. Curr. Biol. 2015, 25, R1158–R1163. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Liu, R.; Wu, J.; Guo, H.; Yao, W.; Li, S.; Lu, Y.; Jia, Y.; Liang, X.; Tang, J.; Zhang, H. Post-translational modifications of histones: Mechanisms, biological functions, and therapeutic targets. MedComm (2020) 2023, 4, e292. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Morgan, M.A.J.; Shilatifard, A. Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 2020, 52, 1271–1281. [Google Scholar] [CrossRef]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef]
- Di Lorenzo, A.; Bedford, M.T. Histone arginine methylation. FEBS Lett. 2011, 585, 2024–2031. [Google Scholar] [CrossRef]
- Dillon, S.C.; Zhang, X.; Trievel, R.C.; Cheng, X. The SET-domain protein superfamily: Protein lysine methyltransferases. Genome Biol. 2005, 6, 227. [Google Scholar] [CrossRef]
- Tsukada, Y.-i.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef]
- Klose, R.J.; Kallin, E.M.; Zhang, Y. JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 2006, 7, 715–727. [Google Scholar] [CrossRef]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef]
- Shioi, A.; Katagi, M.; Okuno, Y.; Mori, K.; Jono, S.; Koyama, H.; Nishizawa, Y. Induction of bone-type alkaline phosphatase in human vascular smooth muscle cells: Roles of tumor necrosis factor-alpha and oncostatin M derived from macrophages. Circ. Res. 2002, 91, 9–16. [Google Scholar] [CrossRef]
- Tintut, Y.; Patel, J.; Parhami, F.; Demer, L.L. Tumor necrosis factor-alpha promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation 2000, 102, 2636–2642. [Google Scholar] [CrossRef]
- Kurozumi, A.; Nakano, K.; Yamagata, K.; Okada, Y.; Nakayamada, S.; Tanaka, Y. IL-6 and sIL-6R induces STAT3-dependent differentiation of human VSMCs into osteoblast-like cells through JMJD2B-mediated histone demethylation of RUNX2. Bone 2019, 124, 53–61. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef]
- Xue, S.; Leng, S.; Zhang, F.; Dang, Z.; Su, G.; Yu, W. Enhancer of zeste homolog 2 facilitates phenotypic transition of vascular smooth muscle cells leading to aortic aneurysm/dissection. Exp. Ther. Med. 2024, 27, 145. [Google Scholar] [CrossRef]
- Cheng, S.L.; Ramachandran, B.; Behrmann, A.; Shao, J.S.; Mead, M.; Smith, C.; Krchma, K.; Bello Arredondo, Y.; Kovacs, A.; Kapoor, K.; et al. Vascular smooth muscle LRP6 limits arteriosclerotic calcification in diabetic LDLR-/- mice by restraining noncanonical Wnt signals. Circ. Res. 2015, 117, 142–156. [Google Scholar] [CrossRef]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef]
- Park, S.-Y.; Kim, J.-S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Pan, X.; Pi, C.; Ruan, X.; Zheng, H.; Zhang, D.; Liu, X. Mammalian Sirtuins and Their Relevance in Vascular Calcification. Front. Pharmacol. 2022, 13, 907835. [Google Scholar] [CrossRef]
- Wang, S.; Hu, S. The Role of Sirtuins in Osteogenic Differentiation of Vascular Smooth Muscle Cells and Vascular Calcification. Front. Cardiovasc. Med. 2022, 9, 894692. [Google Scholar] [CrossRef]
- Phadwal, K.; Feng, D.; Zhu, D.; MacRae, V.E. Autophagy as a novel therapeutic target in vascular calcification. Pharmacol. Ther. 2020, 206, 107430. [Google Scholar] [CrossRef]
- Lino Cardenas, C.L.; Jiang, W.; Kajuluri, L.P.; Singh, K.; Ostrom, K.; Li, R.; Cherbonneau, F.; Boerboom, S.; Birchenough, C.; Roh, K.; et al. Treatment of calcific arterial disease via enhancement of autophagy using GSK343. iScience 2023, 26, 108360. [Google Scholar] [CrossRef]
- Zhou, J.; Zhou, H.; Liu, C.; Huang, L.; Lu, D.; Gao, C. HDAC1-mediated deacetylation of LSD1 regulates vascular calcification by promoting autophagy in chronic renal failure. J. Cell Mol. Med. 2020, 24, 8636–8649. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Liu, P.; Zhang, C.; Huang, Q.; Zhao, Z.; Wu, S.; Li, D.; Liu, H. HDAC2 counteracts vascular calcification by activating autophagy in chronic kidney disease. Faseb J. 2024, 38, e23470. [Google Scholar] [CrossRef]
- Asfaha, Y.; Schrenk, C.; Alves Avelar, L.A.; Hamacher, A.; Pflieger, M.; Kassack, M.U.; Kurz, T. Recent advances in class IIa histone deacetylases research. Bioorg Med. Chem. 2019, 27, 115087. [Google Scholar] [CrossRef] [PubMed]
- Nishino, T.G.; Miyazaki, M.; Hoshino, H.; Miwa, Y.; Horinouchi, S.; Yoshida, M. 14-3-3 regulates the nuclear import of class IIa histone deacetylases. Biochem. Biophys. Res. Commun. 2008, 377, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Abend, A.; Shkedi, O.; Fertouk, M.; Caspi, L.H.; Kehat, I. Salt-inducible kinase induces cytoplasmic histone deacetylase 4 to promote vascular calcification. EMBO Rep. 2017, 18, 1166–1185. [Google Scholar] [CrossRef]
- Chen, Y.; Ju, L.; Rushdi, M.; Ge, C.; Zhu, C. Receptor-mediated cell mechanosensing. Mol. Biol. Cell 2017, 28, 3134–3155. [Google Scholar] [CrossRef]
- Malhotra, R.; Mauer, A.C.; Lino Cardenas, C.L.; Guo, X.; Yao, J.; Zhang, X.; Wunderer, F.; Smith, A.V.; Wong, Q.; Pechlivanis, S.; et al. HDAC9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat. Genet. 2019, 51, 1580–1587. [Google Scholar] [CrossRef]
- Lan, Z.; Chen, A.; Li, L.; Ye, Y.; Liang, Q.; Dong, Q.; Wang, S.; Fu, M.; Li, Y.; Liu, X.; et al. Downregulation of HDAC9 by the ketone metabolite β-hydroxybutyrate suppresses vascular calcification. J. Pathol. 2022, 258, 213–226. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Shi, J.; Yang, Y.; Cheng, A.; Xu, G.; He, F. Metabolism of vascular smooth muscle cells in vascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H613–H631. [Google Scholar] [CrossRef]
- Yang, L.; Gao, L.; Nickel, T.; Yang, J.; Zhou, J.; Gilbertsen, A.; Geng, Z.; Johnson, C.; Young, B.; Henke, C.; et al. Lactate Promotes Synthetic Phenotype in Vascular Smooth Muscle Cells. Circ. Res. 2017, 121, 1251–1262. [Google Scholar] [CrossRef]
- Ma, W.; Jia, K.; Cheng, H.; Xu, H.; Li, Z.; Zhang, H.; Xie, H.; Sun, H.; Yi, L.; Chen, Z.; et al. Orphan Nuclear Receptor NR4A3 Promotes Vascular Calcification via Histone Lactylation. Circ. Res. 2024, 134, 1427–1447. [Google Scholar] [CrossRef] [PubMed]
- Beermann, J.; Piccoli, M.T.; Viereck, J.; Thum, T. Non-coding RNAs in Development and Disease: Background, Mechanisms, and Therapeutic Approaches. Physiol. Rev. 2016, 96, 1297–1325. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Wang, C.; Chen, H. MicroRNAs are critical in regulating smooth muscle cell mineralization and apoptosis during vascular calcification. J. Cell Mol. Med. 2020, 24, 13564–13572. [Google Scholar] [CrossRef]
- Jang, B.; Zhang, D.; Ma, Z.; Yang, X.; Liu, L.; Xing, H.; Feng, L.; Song, J.; Zhao, X.; Song, X.; et al. MicroRNAs in vascular smooth muscle cells: Mechanisms, therapeutic potential, and advances in delivery systems. Life Sci. 2025, 364, 123424. [Google Scholar] [CrossRef] [PubMed]
- Graf, J.; Kretz, M. From structure to function: Route to understanding lncRNA mechanism. Bioessays 2020, 42, e2000027. [Google Scholar] [CrossRef]
- Jeong, G.; Kwon, D.H.; Shin, S.; Choe, N.; Ryu, J.; Lim, Y.H.; Kim, J.; Park, W.J.; Kook, H.; Kim, Y.K. Long noncoding RNAs in vascular smooth muscle cells regulate vascular calcification. Sci. Rep. 2019, 9, 5848. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, J.; Meng, Q.; Li, D.; Hu, F.Z.; Zhu, Y.Q.; Huang, Y.Y.; Liu, Y.N.; Sun, L.; Liang, Q.H. The protective effects of long non-coding RNA-ANCR on arterial calcification. J. Bone Miner. Metab. 2020, 38, 421–431. [Google Scholar] [CrossRef]
- Chang, Z.; Yan, G.; Zheng, J.; Liu, Z. The lncRNA GAS5 Inhibits the Osteogenic Differentiation and Calcification of Human Vascular Smooth Muscle Cells. Calcif. Tissue Int. 2020, 107, 86–95. [Google Scholar] [CrossRef]
- Song, Z.; Wei, D.; Chen, Y.; Chen, L.; Bian, Y.; Shen, Y.; Chen, J.; Pan, Y. Association of astragaloside IV-inhibited autophagy and mineralization in vascular smooth muscle cells with lncRNA H19 and DUSP5-mediated ERK signaling. Toxicol. Appl. Pharmacol. 2019, 364, 45–54. [Google Scholar] [CrossRef]
- Li, X.Z.; Xiong, Z.C.; Zhang, S.L.; Hao, Q.Y.; Liu, Z.Y.; Zhang, H.F.; Wang, J.F.; Gao, J.W.; Liu, P.M. Upregulated LncRNA H19 Sponges MiR-106a-5p and Contributes to Aldosterone-Induced Vascular Calcification via Activating the Runx2-Dependent Pathway. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 1684–1699. [Google Scholar] [CrossRef]
- Wang, T.; Cheng, M.; Jin, J.; Bai, Y.; Zhang, D.; Zhang, S.; Xu, J. Hypomethylation of the LncRNA H19 promoter accelerates osteogenic differentiation of vascular smooth muscle cells by activating the Erk1/2 pathways. J. Int. Med. Res. 2024, 52, 3000605241234567. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Qi, H.; Yao, L. A long non-coding RNA H19/microRNA-138/TLR3 network is involved in high phosphorus-mediated vascular calcification and chronic kidney disease. Cell Cycle 2022, 21, 1667–1683. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Yang, X.C.; Chen, M.L.; Zhuang, Z.W.; Jiang, Y.; Wang, J.; Zhou, Y.J. LncRNA H19/Runx2 axis promotes VSMCs transition via MAPK pathway. Am. J. Transl. Res. 2020, 12, 1338–1347. [Google Scholar] [PubMed]
- Gong, Y.; Zhong, Q.; Xia, Y.; Wen, Y.; Gan, H. Long non-coding RNA MALAT1 sponges miR-30c to promote the calcification of human vascular smooth muscle cells by regulating Runx2. Ren. Fail. 2023, 45, 2204953. [Google Scholar] [CrossRef]
- Lin, X.; Zhan, J.K.; Zhong, J.Y.; Wang, Y.J.; Wang, Y.; Li, S.; He, J.Y.; Tan, P.; Chen, Y.Y.; Liu, X.B.; et al. lncRNA-ES3/miR-34c-5p/BMF axis is involved in regulating high-glucose-induced calcification/senescence of VSMCs. Aging 2019, 11, 523–535. [Google Scholar] [CrossRef]
- Li, S.; Ni, Y.; Li, C.; Xiang, Q.; Zhao, Y.; Xu, H.; Huang, W.; Wang, Y.; Wang, Y.; Zhan, J.; et al. Long noncoding RNA SNHG1 alleviates high glucose-induced vascular smooth muscle cells calcification/senescence by post-transcriptionally regulating Bhlhe40 and autophagy via Atg10. J. Physiol. Biochem. 2023, 79, 83–105. [Google Scholar] [CrossRef]
- Zhong, J.Y.; Cui, X.J.; Zhan, J.K.; Wang, Y.J.; Li, S.; Lin, X.; Xiang, Q.Y.; Ni, Y.Q.; Liu, L.; Liu, Y.S. LncRNA-ES3 inhibition by Bhlhe40 is involved in high glucose-induced calcification/senescence of vascular smooth muscle cells. Ann. N. Y. Acad. Sci. 2020, 1474, 61–72. [Google Scholar] [CrossRef]
- Huang, C.; Zhan, J.F.; Chen, Y.X.; Xu, C.Y.; Chen, Y. LncRNA-SNHG29 inhibits vascular smooth muscle cell calcification by downregulating miR-200b-3p to activate the α-Klotho/FGFR1/FGF23 axis. Cytokine 2020, 136, 155243. [Google Scholar] [CrossRef]
- Li, Y.; Xi, Z.; Yu, Z.; Yang, C.; Tan, C. LincRNA-EPS increases TGF-β expression to inhibit the Wnt/β-catenin pathway, VSMC osteoblastic differentiation and vascular calcification in diabetic mice. Exp. Ther. Med. 2022, 23, 425. [Google Scholar] [CrossRef]
- Horita, H.; Wysoczynski, C.L.; Walker, L.A.; Moulton, K.S.; Li, M.; Ostriker, A.; Tucker, R.; McKinsey, T.A.; Churchill, M.E.; Nemenoff, R.A.; et al. Nuclear PTEN functions as an essential regulator of SRF-dependent transcription to control smooth muscle differentiation. Nat. Commun. 2016, 7, 10830. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Kontos, C.D. Inhibition of vascular smooth muscle cell proliferation, migration, and survival by the tumor suppressor protein PTEN. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Guo, Y.; Diao, Z.; Guo, W.; Liu, W. Genome-wide identification of lncRNAs and mRNAs differentially expressed in human vascular smooth muscle cells stimulated by high phosphorus. Ren. Fail. 2020, 42, 437–446. [Google Scholar] [CrossRef]
- Wicik, Z.; Jales Neto, L.H.; Guzman, L.E.F.; Pavão, R.; Takayama, L.; Caparbo, V.F.; Lopes, N.H.M.; Pereira, A.C.; Pereira, R.M.R. The crosstalk between bone metabolism, lncRNAs, microRNAs and mRNAs in coronary artery calcification. Genomics 2021, 113, 503–513. [Google Scholar] [CrossRef]
- Wei, X.; Su, Y.; Li, Q.; Zheng, Z.; Hou, P. Analysis of crucial genes, pathways and construction of the molecular regulatory networks in vascular smooth muscle cell calcification. Exp. Ther. Med. 2021, 21, 589. [Google Scholar] [CrossRef]
- Goettsch, C.; Hutcheson, J.D.; Aikawa, E. MicroRNA in cardiovascular calcification: Focus on targets and extracellular vesicle delivery mechanisms. Circ. Res. 2013, 112, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xiao, X.; Shen, Y.; Chen, L.; Xu, C.; Zhao, H.; Wu, Y.; Zhang, Q.; Zhong, J.; Tang, Z.; et al. MicroRNA-32 promotes calcification in vascular smooth muscle cells: Implications as a novel marker for coronary artery calcification. PLoS ONE 2017, 12, e0174138. [Google Scholar] [CrossRef]
- Guo, X.; Liu, S.; Wu, X.; Yang, R.; Ren, Q.; Zhou, Y.; Shi, K.; Yuan, L.; Zhang, N.; Liu, S. Alleviating vascular calcification with Bushen Huoxue formula in rats with chronic kidney disease by inhibiting the PTEN/PI3K/AKT signaling pathway through exosomal microRNA-32. J. Pharm. Pharmacol. 2025, 77, 550–563. [Google Scholar] [CrossRef]
- Cao, J.; Chen, C.; Chen, Q.; Gao, Y.; Zhao, Z.; Yuan, Q.; Li, A.; Yang, S.; He, Y.; Zu, X.; et al. Extracellular vesicle miR-32 derived from macrophage promotes arterial calcification in mice with type 2 diabetes via inhibiting VSMC autophagy. J. Transl. Med. 2022, 20, 307. [Google Scholar] [CrossRef]
- Cao, J.; Chen, L.; Zhong, X.; Shen, Y.; Gao, Y.; Chen, Q.; Zu, X.; Liu, J. miR32-5p promoted vascular smooth muscle cell calcification by upregulating TNFα in the microenvironment. BMC Immunol. 2020, 21, 3. [Google Scholar] [CrossRef]
- Zuccolo, E.; Badi, I.; Scavello, F.; Gambuzza, I.; Mancinelli, L.; Macrì, F.; Tedesco, C.C.; Veglia, F.; Bonfigli, A.R.; Olivieri, F.; et al. The microRNA-34a-Induced Senescence-Associated Secretory Phenotype (SASP) Favors Vascular Smooth Muscle Cells Calcification. Int. J. Mol. Sci. 2020, 21, 4454. [Google Scholar] [CrossRef] [PubMed]
- Badi, I.; Mancinelli, L.; Polizzotto, A.; Ferri, D.; Zeni, F.; Burba, I.; Milano, G.; Brambilla, F.; Saccu, C.; Bianchi, M.E.; et al. miR-34a Promotes Vascular Smooth Muscle Cell Calcification by Downregulating SIRT1 (Sirtuin 1) and Axl (AXL Receptor Tyrosine Kinase). Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wang, Z.; Wei, L.; Cheng, X.; Chen, L.; Gao, F.; Jiang, H. Indoxyl sulfate promotes osteogenic differentiation of vascular smooth muscle cells by miR-155-5p-dependent downregulation of matrix Gla protein via ROS/NF-κB signaling. Exp. Cell Res. 2020, 397, 112301. [Google Scholar] [CrossRef]
- Li, Y.; Sun, W.; Saaoud, F.; Wang, Y.; Wang, Q.; Hodge, J.; Hui, Y.; Yin, S.; Lessner, S.M.; Kong, X.; et al. MiR155 modulates vascular calcification by regulating Akt-FOXO3a signalling and apoptosis in vascular smooth muscle cells. J. Cell Mol. Med. 2021, 25, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Ning, H.; Wang, X.; Wang, Y.; Han, T.; Sun, C. Endothelial Autophagy Promotes Atheroprotective Communication Between Endothelial and Smooth Muscle Cells via Exosome-Mediated Delivery of miR-204-5p. Arterioscler. Thromb. Vasc. Biol. 2024, 44, 1813–1832. [Google Scholar] [CrossRef]
- Liao, X.B.; Zhang, Z.Y.; Yuan, K.; Liu, Y.; Feng, X.; Cui, R.R.; Hu, Y.R.; Yuan, Z.S.; Gu, L.; Li, S.J.; et al. MiR-133a modulates osteogenic differentiation of vascular smooth muscle cells. Endocrinology 2013, 154, 3344–3352. [Google Scholar] [CrossRef]
- Kaul, A.; Dhalla, P.S.; Bapatla, A.; Khalid, R.; Garcia, J.; Armenta-Quiroga, A.S.; Khan, S. Current Treatment Modalities for Calcified Coronary Artery Disease: A Review Article Comparing Novel Intravascular Lithotripsy and Traditional Rotational Atherectomy. Cureus 2020, 12, e10922. [Google Scholar] [CrossRef]
- Brinton, T.J.; Ali, Z.A.; Hill, J.M.; Meredith, I.T.; Maehara, A.; Illindala, U.; Lansky, A.; Götberg, M.; Van Mieghem, N.M.; Whitbourn, R.; et al. Feasibility of Shockwave Coronary Intravascular Lithotripsy for the Treatment of Calcified Coronary Stenoses. Circulation 2019, 139, 834–836. [Google Scholar] [CrossRef]
- Murali, S.; Smith, E.R.; Tiong, M.K.; Tan, S.J.; Toussaint, N.D. Interventions to Attenuate Cardiovascular Calcification Progression: A Systematic Review of Randomized Clinical Trials. J. Am. Heart Assoc. 2023, 12, e031676. [Google Scholar] [CrossRef]
- Campbell, J.H.; Efendy, J.L.; Smith, N.J.; Campbell, G.R. Molecular basis by which garlic suppresses atherosclerosis. J. Nutr. 2001, 131, 1006s–1009s. [Google Scholar] [CrossRef]
- Budoff, M.J.; Ahmadi, N.; Gul, K.M.; Liu, S.T.; Flores, F.R.; Tiano, J.; Takasu, J.; Miller, E.; Tsimikas, S. Aged garlic extract supplemented with B vitamins, folic acid and L-arginine retards the progression of subclinical atherosclerosis: A randomized clinical trial. Prev. Med. 2009, 49, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Budoff, M.J.; Takasu, J.; Flores, F.R.; Niihara, Y.; Lu, B.; Lau, B.H.; Rosen, R.T.; Amagase, H. Inhibiting progression of coronary calcification using Aged Garlic Extract in patients receiving statin therapy: A preliminary study. Prev. Med. 2004, 39, 985–991. [Google Scholar] [CrossRef]
- Zeb, I.; Ahmadi, N.; Nasir, K.; Kadakia, J.; Larijani, V.N.; Flores, F.; Li, D.; Budoff, M.J. Aged garlic extract and coenzyme Q10 have favorable effect on inflammatory markers and coronary atherosclerosis progression: A randomized clinical trial. J. Cardiovasc. Dis. Res. 2012, 3, 185–190. [Google Scholar] [CrossRef]
- Wlosinska, M.; Nilsson, A.C.; Hlebowicz, J.; Hauggaard, A.; Kjellin, M.; Fakhro, M.; Lindstedt, S. The effect of aged garlic extract on the atherosclerotic process—A randomized double-blind placebo-controlled trial. BMC Complement. Med. Ther. 2020, 20, 132. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Nakanishi, R.; Li, D.; Alani, A.; Rezaeian, P.; Prabhu, S.; Abraham, J.; Fahmy, M.A.; Dailing, C.; Flores, F.; et al. Aged Garlic Extract Reduces Low Attenuation Plaque in Coronary Arteries of Patients with Metabolic Syndrome in a Prospective Randomized Double-Blind Study. J. Nutr. 2016, 146, 427s–432s. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, K.; Kinninger, A.; Cherukuri, L.; Birudaraju, D.; Nakanishi, R.; Almeida, S.; Jayawardena, E.; Shekar, C.; Flores, F.; Hamal, S.; et al. Aged garlic extract reduces low attenuation plaque in coronary arteries of patients with diabetes: A randomized, double-blind, placebo-controlled study. Exp. Ther. Med. 2020, 19, 1457–1461. [Google Scholar] [CrossRef]
- Arad, Y.; Spadaro, L.A.; Roth, M.; Newstein, D.; Guerci, A.D. Treatment of asymptomatic adults with elevated coronary calcium scores with atorvastatin, vitamin C, and vitamin E: The St. Francis Heart Study randomized clinical trial. J. Am. Coll. Cardiol. 2005, 46, 166–172. [Google Scholar] [CrossRef]
- Cowell, S.J.; Newby, D.E.; Prescott, R.J.; Bloomfield, P.; Reid, J.; Northridge, D.B.; Boon, N.A. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N. Engl. J. Med. 2005, 352, 2389–2397. [Google Scholar] [CrossRef]
- Dichtl, W.; Alber, H.F.; Feuchtner, G.M.; Hintringer, F.; Reinthaler, M.; Bartel, T.; Süssenbacher, A.; Grander, W.; Ulmer, H.; Pachinger, O.; et al. Prognosis and risk factors in patients with asymptomatic aortic stenosis and their modulation by atorvastatin (20 mg). Am. J. Cardiol. 2008, 102, 743–748. [Google Scholar] [CrossRef]
- Houslay, E.S.; Cowell, S.J.; Prescott, R.J.; Reid, J.; Burton, J.; Northridge, D.B.; Boon, N.A.; Newby, D.E. Progressive coronary calcification despite intensive lipid-lowering treatment: A randomised controlled trial. Heart 2006, 92, 1207–1212. [Google Scholar] [CrossRef]
- Miyoshi, T.; Kohno, K.; Asonuma, H.; Sakuragi, S.; Nakahama, M.; Kawai, Y.; Uesugi, T.; Oka, T.; Munemasa, M.; Takahashi, N.; et al. Effect of Intensive and Standard Pitavastatin Treatment With or Without Eicosapentaenoic Acid on Progression of Coronary Artery Calcification over 12 Months—Prospective Multicenter Study. Circ. J. 2018, 82, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.A.; Kiani, A.N.; Post, W.; Christopher-Stine, L.; Magder, L.S. Lupus Atherosclerosis Prevention Study (LAPS). Ann. Rheum. Dis. 2011, 70, 760–765. [Google Scholar] [CrossRef]
- Longenecker, C.T.; Sattar, A.; Gilkeson, R.; McComsey, G.A. Rosuvastatin slows progression of subclinical atherosclerosis in patients with treated HIV infection. Aids 2016, 30, 2195–2203. [Google Scholar] [CrossRef]
- Raggi, P.; Callister, T.Q.; Davidson, M.; Welty, F.K.; Bachmann, G.A.; Laskey, R.; Pittman, D.; Kafonek, S.; Scott, R. Aggressive versus moderate lipid-lowering therapy in postmenopausal women with hypercholesterolemia: Rationale and design of the Beyond Endorsed Lipid Lowering with EBT Scanning (BELLES) trial. Am. Heart J. 2001, 141, 722–726. [Google Scholar] [CrossRef]
- Schmermund, A.; Achenbach, S.; Budde, T.; Buziashvili, Y.; Förster, A.; Friedrich, G.; Henein, M.; Kerkhoff, G.; Knollmann, F.; Kukharchuk, V.; et al. Effect of intensive versus standard lipid-lowering treatment with atorvastatin on the progression of calcified coronary atherosclerosis over 12 months: A multicenter, randomized, double-blind trial. Circulation 2006, 113, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Terry, J.G.; Carr, J.J.; Kouba, E.O.; Davis, D.H.; Menon, L.; Bender, K.; Chandler, E.T.; Morgan, T.; Crouse, J.R., 3rd. Effect of simvastatin (80 mg) on coronary and abdominal aortic arterial calcium (from the coronary artery calcification treatment with zocor [CATZ] study). Am. J. Cardiol. 2007, 99, 1714–1717. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Kang, S.J.; Ahn, J.M.; Chang, M.; Yun, S.C.; Roh, J.H.; Lee, P.H.; Park, H.W.; Yoon, S.H.; Park, D.W.; et al. Effect of Statin Treatment on Modifying Plaque Composition: A Double-Blind, Randomized Study. J. Am. Coll. Cardiol. 2016, 67, 1772–1783. [Google Scholar] [CrossRef]
- Egede, R.; Jensen, L.O.; Hansen, H.S.; Hansen, K.N.; Junker, A.; Thayssen, P. Influence of high-dose lipid lowering treatment compared to low-dose lipid lowering treatment on plaque composition assessed by intravascular ultrasound virtual histology in patients with ST-segment elevation acute myocardial infarction: The VIRHISTAMI trial. EuroIntervention 2013, 8, 1182–1189. [Google Scholar] [CrossRef]
- Lo, J.; Lu, M.T.; Ihenachor, E.J.; Wei, J.; Looby, S.E.; Fitch, K.V.; Oh, J.; Zimmerman, C.O.; Hwang, J.; Abbara, S.; et al. Effects of statin therapy on coronary artery plaque volume and high-risk plaque morphology in HIV-infected patients with subclinical atherosclerosis: A randomised, double-blind, placebo-controlled trial. Lancet HIV 2015, 2, e52–e63. [Google Scholar] [CrossRef]
- Ayombil, F.; Camire, R.M. Insights into vitamin K-dependent carboxylation: Home field advantage. Haematologica 2020, 105, 1996–1998. [Google Scholar] [CrossRef]
- Yao, Y.; Shahbazian, A.; Boström, K.I. Proline and γ-Carboxylated Glutamate Residues in Matrix Gla Protein Are Critical for Binding of Bone Morphogenetic Protein-4. Circ. Res. 2008, 102, 1065–1074. [Google Scholar] [CrossRef]
- Shea, M.K.; O’Donnell, C.J.; Hoffmann, U.; Dallal, G.E.; Dawson-Hughes, B.; Ordovas, J.M.; Price, P.A.; Williamson, M.K.; Booth, S.L. Vitamin K supplementation and progression of coronary artery calcium in older men and women. Am. J. Clin. Nutr. 2009, 89, 1799–1807. [Google Scholar] [CrossRef]
- Bellinge, J.W.; Francis, R.J.; Lee, S.C.; Bondonno, N.P.; Sim, M.; Lewis, J.R.; Watts, G.F.; Schultz, C.J. The effect of vitamin K1 on arterial calcification activity in subjects with diabetes mellitus: A post hoc analysis of a double-blind, randomized, placebo-controlled trial. Am. J. Clin. Nutr. 2022, 115, 45–52. [Google Scholar] [CrossRef]
- Henzel, J.; Kępka, C.; Kruk, M.; Makarewicz-Wujec, M.; Wardziak, Ł.; Trochimiuk, P.; Dzielińska, Z.; Demkow, M. High-Risk Coronary Plaque Regression After Intensive Lifestyle Intervention in Nonobstructive Coronary Disease: A Randomized Study. JACC Cardiovasc. Imaging 2021, 14, 1192–1202. [Google Scholar] [CrossRef]
- Fitch, K.; Abbara, S.; Lee, H.; Stavrou, E.; Sacks, R.; Michel, T.; Hemphill, L.; Torriani, M.; Grinspoon, S. Effects of lifestyle modification and metformin on atherosclerotic indices among HIV-infected patients with the metabolic syndrome. Aids 2012, 26, 587–597. [Google Scholar] [CrossRef]
- Kuller, L.H.; Pettee Gabriel, K.K.; Kinzel, L.S.; Underwood, D.A.; Conroy, M.B.; Chang, Y.; Mackey, R.H.; Edmundowicz, D.; Tyrrell, K.S.; Buhari, A.M.; et al. The Women on the Move Through Activity and Nutrition (WOMAN) study: Final 48-month results. Obesity 2012, 20, 636–643. [Google Scholar] [CrossRef]
- Lehmann, N.; Paul, A.; Moebus, S.; Budde, T.; Dobos, G.J.; Michalsen, A. Effects of lifestyle modification on coronary artery calcium progression and prognostic factors in coronary patients--3-year results of the randomized SAFE-LIFE trial. Atherosclerosis 2011, 219, 630–636. [Google Scholar] [CrossRef]
- Motro, M.; Shemesh, J. Calcium channel blocker nifedipine slows down progression of coronary calcification in hypertensive patients compared with diuretics. Hypertension 2001, 37, 1410–1413. [Google Scholar] [CrossRef]
- Motro, M.; Kirwan, B.A.; de Brouwer, S.; Poole-Wilson, P.A.; Shemesh, J. Tracking coronary calcification and atherosclerotic lesions in patients with stable angina pectoris undergoing nifedipine therapy. Cardiology 2007, 107, 165–171. [Google Scholar] [CrossRef]
- Budoff, M.J.; Muhlestein, J.B.; Bhatt, D.L.; Le Pa, V.T.; May, H.T.; Shaikh, K.; Shekar, C.; Kinninger, A.; Lakshmanan, S.; Roy, S.K.; et al. Effect of icosapent ethyl on progression of coronary atherosclerosis in patients with elevated triglycerides on statin therapy: A prospective, placebo-controlled randomized trial (EVAPORATE): Interim results. Cardiovasc. Res. 2021, 117, 1070–1077. [Google Scholar] [CrossRef]
- Alfaddagh, A.; Elajami, T.K.; Ashfaque, H.; Saleh, M.; Bistrian, B.R.; Welty, F.K. Effect of Eicosapentaenoic and Docosahexaenoic Acids Added to Statin Therapy on Coronary Artery Plaque in Patients With Coronary Artery Disease: A Randomized Clinical Trial. J. Am. Heart Assoc. 2017, 6, 6981. [Google Scholar] [CrossRef]
- Davidson, M.H.; Beam, C.A.; Haffner, S.; Perez, A.; D’Agostino, R., Sr.; Mazzone, T. Pioglitazone versus glimepiride on coronary artery calcium progression in patients with type 2 diabetes mellitus: A secondary end point of the CHICAGO study. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1873–1876. [Google Scholar] [CrossRef]
- Nozue, T.; Fukui, K.; Koyama, Y.; Fujii, H.; Kunishima, T.; Hikita, H.; Hibi, K.; Miyazawa, A.; Michishita, I. Effects of sitagliptin on coronary atherosclerosis in patients with type 2 diabetes-A serial integrated backscatter-intravascular ultrasound study. Am. J. Cardiovasc. Dis. 2016, 6, 153–162. [Google Scholar]
- Basaria, S.; Harman, S.M.; Travison, T.G.; Hodis, H.; Tsitouras, P.; Budoff, M.; Pencina, K.M.; Vita, J.; Dzekov, C.; Mazer, N.A.; et al. Effects of Testosterone Administration for 3 Years on Subclinical Atherosclerosis Progression in Older Men With Low or Low-Normal Testosterone Levels: A Randomized Clinical Trial. Jama 2015, 314, 570–581. [Google Scholar] [CrossRef]
- Budoff, M.J.; Ellenberg, S.S.; Lewis, C.E.; Mohler, E.R., 3rd; Wenger, N.K.; Bhasin, S.; Barrett-Connor, E.; Swerdloff, R.S.; Stephens-Shields, A.; Cauley, J.A.; et al. Testosterone Treatment and Coronary Artery Plaque Volume in Older Men With Low Testosterone. Jama 2017, 317, 708–716. [Google Scholar] [CrossRef]
- Harman, S.M.; Black, D.M.; Naftolin, F.; Brinton, E.A.; Budoff, M.J.; Cedars, M.I.; Hopkins, P.N.; Lobo, R.A.; Manson, J.E.; Merriam, G.R.; et al. Arterial imaging outcomes and cardiovascular risk factors in recently menopausal women: A randomized trial. Ann. Intern. Med. 2014, 161, 249–260. [Google Scholar] [CrossRef]
- Lee, D.H.; Chun, E.J.; Hur, J.H.; Min, S.H.; Lee, J.E.; Oh, T.J.; Kim, K.M.; Jang, H.C.; Han, S.J.; Kang, D.K.; et al. Effect of sarpogrelate, a selective 5-HT(2A) receptor antagonist, on characteristics of coronary artery disease in patients with type 2 diabetes. Atherosclerosis 2017, 257, 47–54. [Google Scholar] [CrossRef]
- Lee, D.H.; Chun, E.J.; Oh, T.J.; Kim, K.M.; Moon, J.H.; Choi, S.H.; Park, K.S.; Jang, H.C.; Lim, S. Effect of cilostazol, a phosphodiesterase-3 inhibitor, on coronary artery stenosis and plaque characteristics in patients with type 2 diabetes: ESCAPE study. Diabetes Obes. Metab. 2019, 21, 1409–1418. [Google Scholar] [CrossRef]
- Kaminskas, E.; Farrell, A.T.; Wang, Y.C.; Sridhara, R.; Pazdur, R. FDA drug approval summary: Azacitidine (5-azacytidine, Vidaza) for injectable suspension. Oncologist 2005, 10, 176–182. [Google Scholar] [CrossRef]
- Straining, R.; Eighmy, W. Tazemetostat: EZH2 Inhibitor. J. Adv. Pract. Oncol. 2022, 13, 158–163. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A. Discovery and development of SAHA as an anticancer agent. Oncogene 2007, 26, 1351–1356. [Google Scholar] [CrossRef]
- Grant, C.; Rahman, F.; Piekarz, R.; Peer, C.; Frye, R.; Robey, R.W.; Gardner, E.R.; Figg, W.D.; Bates, S.E. Romidepsin: A new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert. Rev. Anticancer. Ther. 2010, 10, 997–1008. [Google Scholar] [CrossRef]
- El Omari, N.; Bakrim, S.; Khalid, A.; Abdalla, A.N.; Almalki, W.H.; Lee, L.H.; Ardianto, C.; Ming, L.C.; Bouyahya, A. Molecular mechanisms underlying the clinical efficacy of panobinostat involve Stochasticity of epigenetic signaling, sensitization to anticancer drugs, and induction of cellular cell death related to cellular stresses. Biomed. Pharmacother. 2023, 164, 114886. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.; Thomas, C.M. Belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. J. Oncol. Pharm. Pract. 2017, 23, 143–147. [Google Scholar] [CrossRef]
- Yu, C.; Li, L.; Xie, F.; Guo, S.; Liu, F.; Dong, N.; Wang, Y. LncRNA TUG1 sponges miR-204-5p to promote osteoblast differentiation through upregulating Runx2 in aortic valve calcification. Cardiovasc. Res. 2018, 114, 168–179. [Google Scholar] [CrossRef]
- Wu, J.; Wan, M.; Jiang, Z.; Gong, W.; Zhou, X. lncRNA FAS-AS1 served as a diagnostic biomarker of end-stage renal disease and mediated vascular calcification via regulating oxidative stress and inflammation. Gene 2024, 896, 148035. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Xiao, H.; Zhou, X.; Li, Y.; Zhao, S.; Zhao, X.; Liu, Y.; Liu, M.; Xue, F.; Zhang, Q.; et al. Engineered exosomes reprogram Gli1(+) cells in vivo to prevent calcification of vascular grafts and autologous pathological vessels. Sci. Adv. 2023, 9, eadf7858. [Google Scholar] [CrossRef]
- Ratti, M.; Lampis, A.; Ghidini, M.; Salati, M.; Mirchev, M.B.; Valeri, N.; Hahne, J.C. MicroRNAs (miRNAs) and Long Non-Coding RNAs (lncRNAs) as New Tools for Cancer Therapy: First Steps from Bench to Bedside. Target. Oncol. 2020, 15, 261–278. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kajuluri, L.P.; Guo, Y.Y.; Lee, S.; Christof, M.; Malhotra, R. Epigenetic Regulation of Human Vascular Calcification. Genes 2025, 16, 506. https://doi.org/10.3390/genes16050506
Kajuluri LP, Guo YY, Lee S, Christof M, Malhotra R. Epigenetic Regulation of Human Vascular Calcification. Genes. 2025; 16(5):506. https://doi.org/10.3390/genes16050506
Chicago/Turabian StyleKajuluri, Lova Prasadareddy, Yugene Young Guo, Sujin Lee, Michael Christof, and Rajeev Malhotra. 2025. "Epigenetic Regulation of Human Vascular Calcification" Genes 16, no. 5: 506. https://doi.org/10.3390/genes16050506
APA StyleKajuluri, L. P., Guo, Y. Y., Lee, S., Christof, M., & Malhotra, R. (2025). Epigenetic Regulation of Human Vascular Calcification. Genes, 16(5), 506. https://doi.org/10.3390/genes16050506