
Whole-Genome Insights into the Genetic Basis of Conformation Traits in German Black Pied (DSN) Cattle


Abstract
1. Introduction
2. Materials and Methods
2.1. Population and Phenotypes
2.2. Genotypes
2.3. GWAS Model
2.4. Significant Threshold and QTL Definition
2.5. QTL Annotation
3. Results
3.1. Correlation Analysis
3.2. Overview of GWAS Results for All Composite Scores and Linear Traits
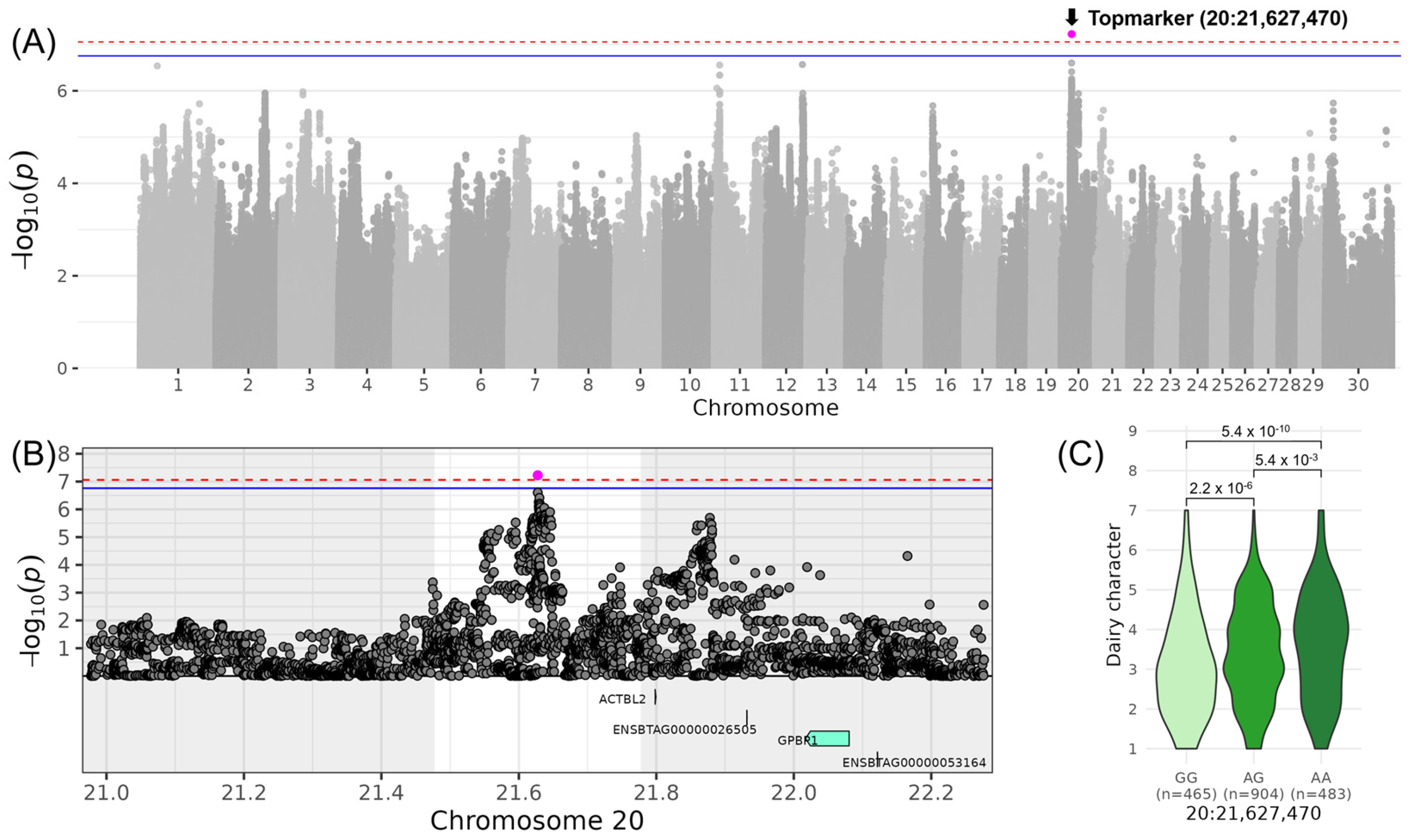
3.3. DAIRY STRENGTH—Association for “Dairy Character”
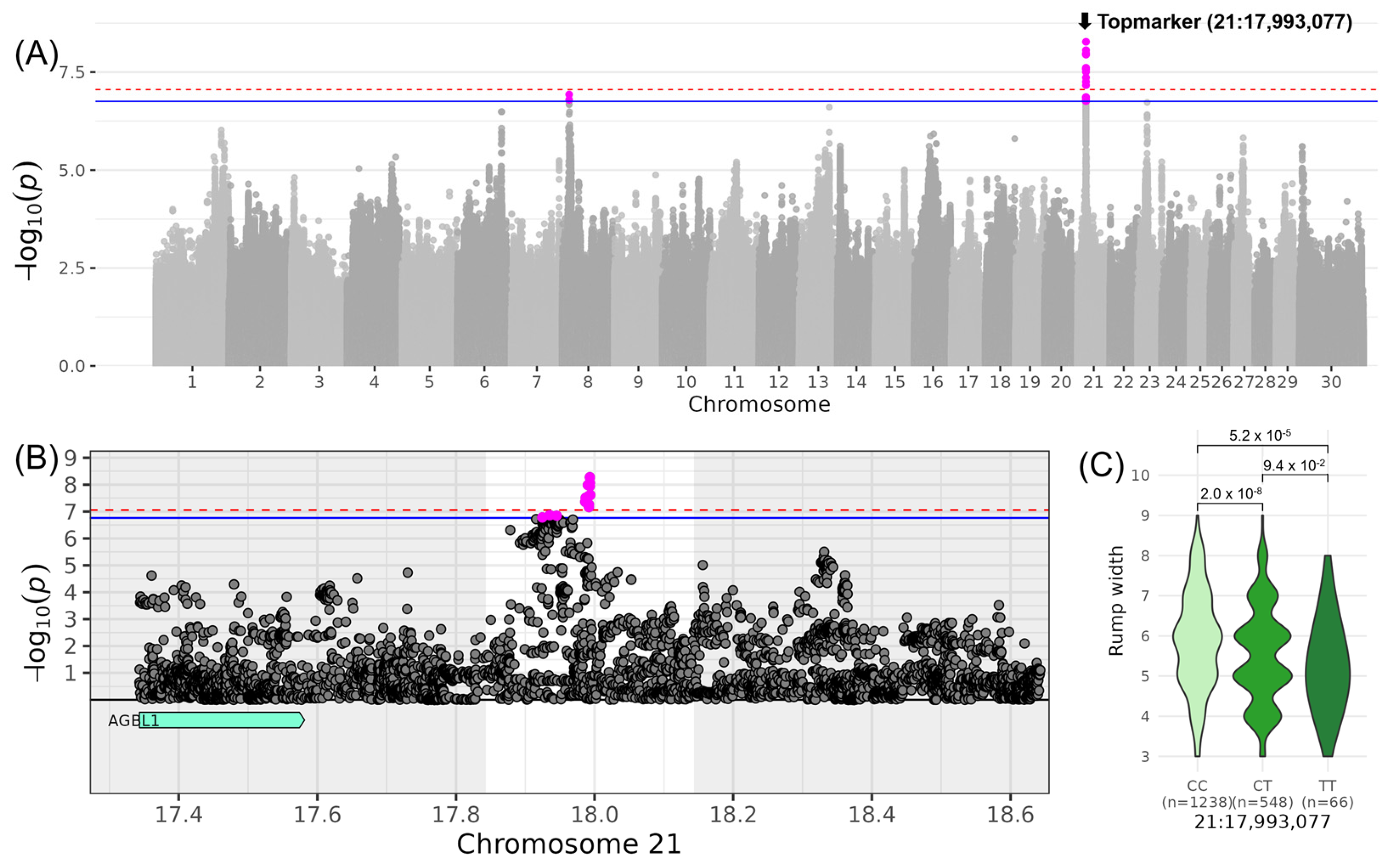
3.4. BODY CONFORMATION—Associations for “Rump Width” and “Rump Angle”
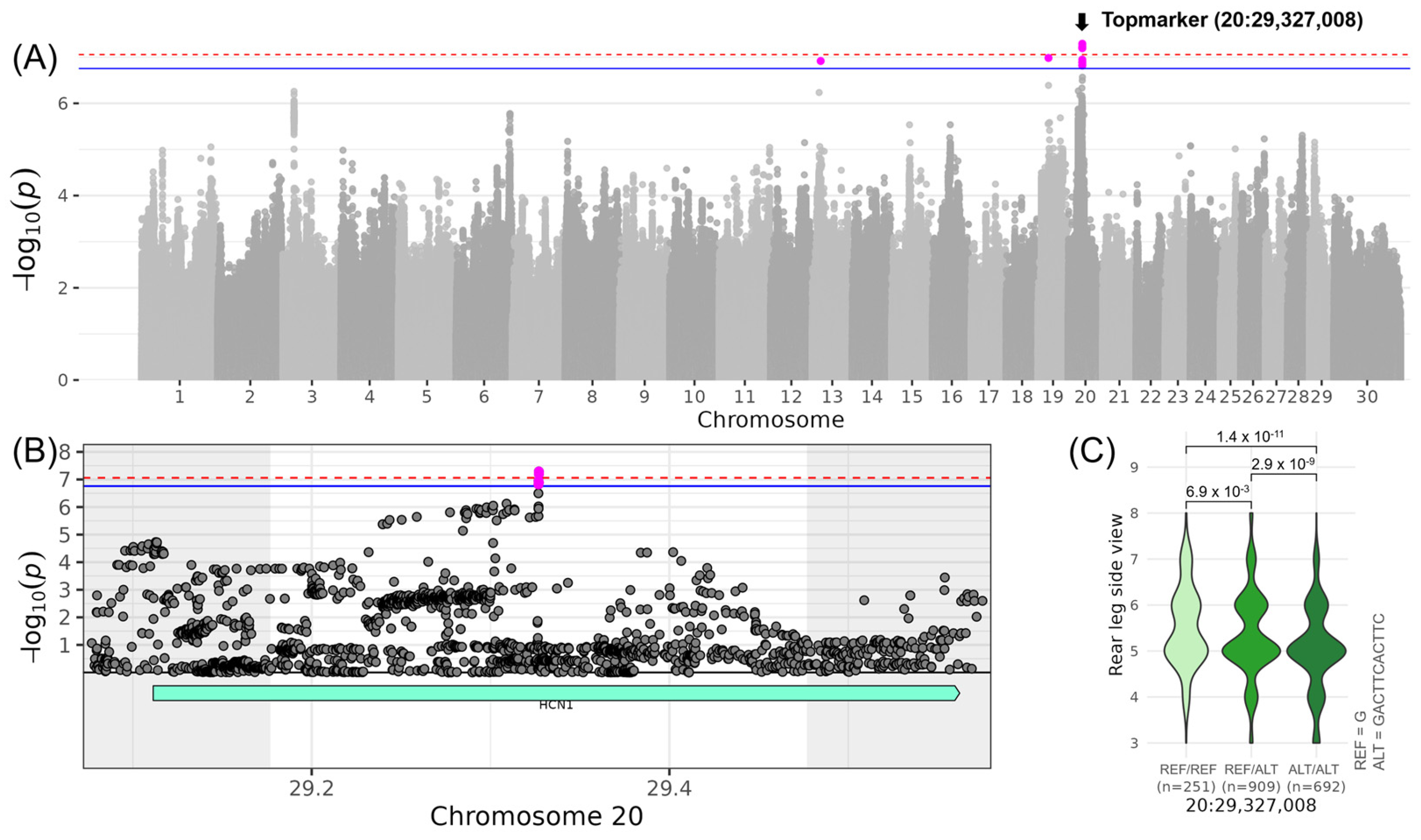
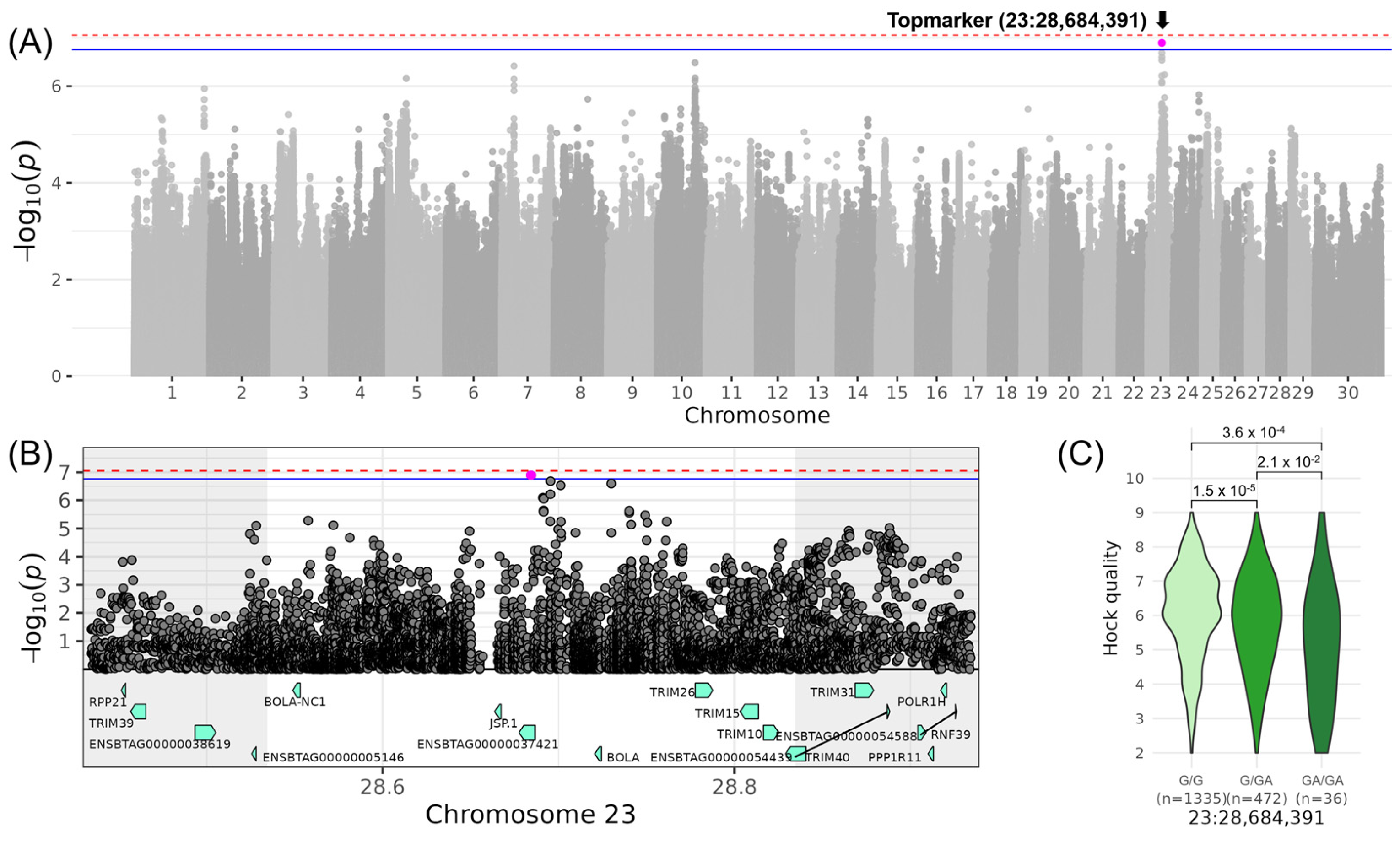
3.5. LEGS&FEET—Associations for “Rear Leg Side” and “Hock Quality”
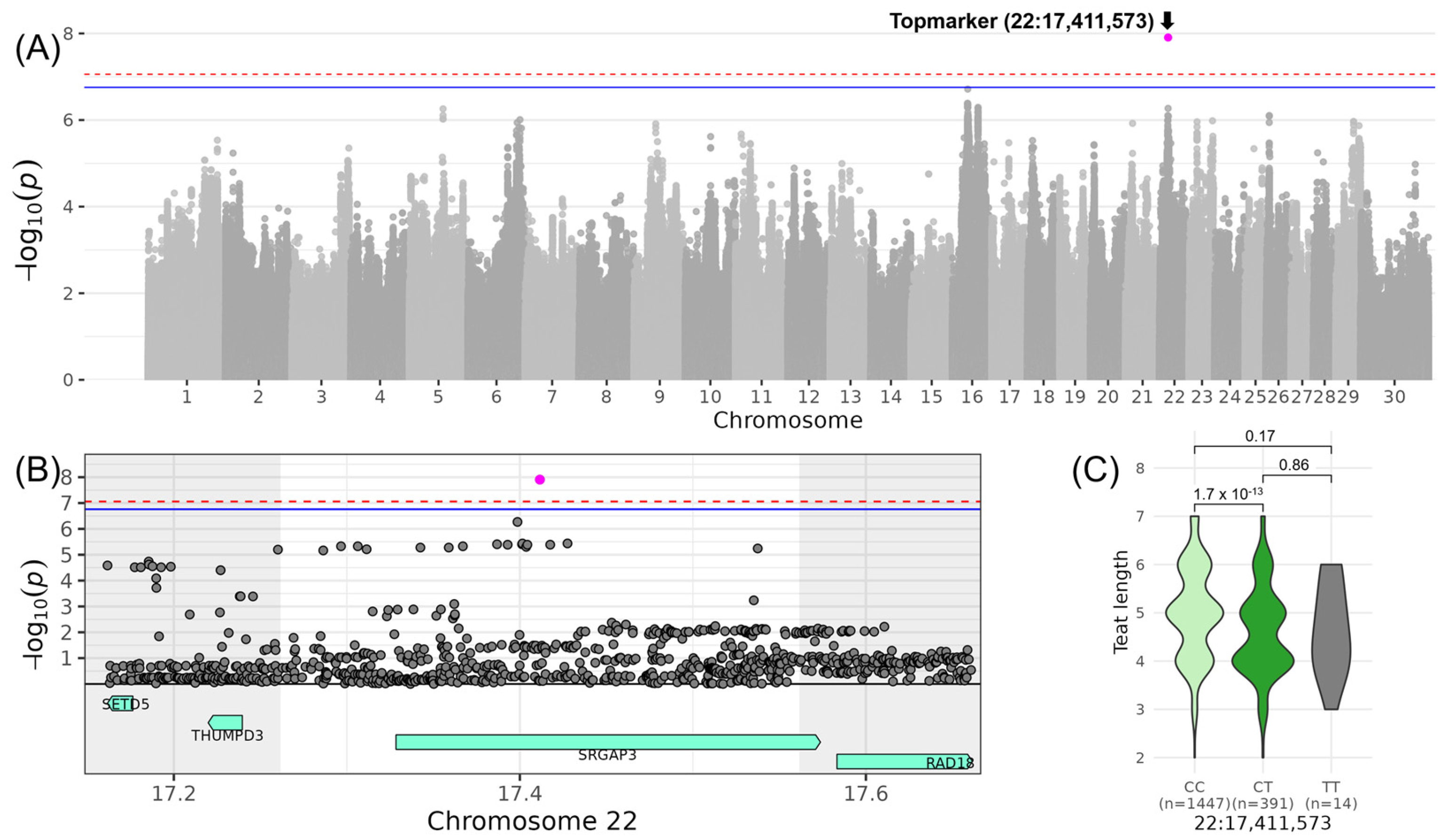
3.6. MAMMARY—Associations for “Central Ligament” and “Teat Length”
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bundesanstalt für Landwirtschaft und Ernährung (BLE). Einheimische Nutztierrassen in Deutschland Und Rote Liste Gefährdeter Nutztierrassen 2023; BLE: Bonn, Germany, 2023. [Google Scholar]
- Adler, B.; Gassan, M.; Thiele, M. Deutsches Schwarzbuntes Niederungsrind—50 Jahre DSN-Genreservezucht in Brandenburg; RBB Rinderproduktion Berlin-Brandenburg GmbH: Groß Kreutz, Germany, 2023. [Google Scholar]
- Rinderzuchtverband Berlin-Brandenburg eG (RZB eG). Zuchtprogramm Für Die Rasse Deutsches Schwarzbuntes Niederungsrind; RZB eG: Groß Kreutz, Germany, 2022. [Google Scholar]
- Pausch, H.; Emmerling, R.; Schwarzenbacher, H.; Fries, R. A Multi-Trait Meta-Analysis with Imputed Sequence Variants Reveals Twelve QTL for Mammary Gland Morphology in Fleckvieh Cattle. Genet. Sel. Evol. GSE 2016, 48, 14. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Fang, M.; Liu, L.; Wang, S.; Liu, J.; Ding, X.; Zhang, S.; Zhang, Q.; Zhang, Y.; Qiao, L.; et al. Genome Wide Association Studies for Body Conformation Traits in the Chinese Holstein Cattle Population. BMC Genom. 2013, 14, 897. [Google Scholar] [CrossRef] [PubMed]
- Flury, C.; Boschung, C.; Denzler, M.; Bapst, B.; Schnyder, U.; Gredler, B.; Signer-Hasler, H. Genome-wide association study for 13 udder traits from linear type classification in cattle. In Proceedings of the 10th World Congress on Genetics Applied to Livestock Production, Vancouver, BC, Canada, 17–22 August 2014. [Google Scholar] [CrossRef]
- Nazar, M.; Abdalla, I.M.; Chen, Z.; Ullah, N.; Liang, Y.; Chu, S.; Xu, T.; Mao, Y.; Yang, Z.; Lu, X. Genome-Wide Association Study for Udder Conformation Traits in Chinese Holstein Cattle. Animals 2022, 12, 2542. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Yin, T.; Neumann, G.B.; Korkuć, P.; Brockmann, G.A.; König, S.; May, K. Genome-Wide Association Study Using Whole-Genome Sequence Data for Fertility, Health Indicator, and Endoparasite Infection Traits in German Black Pied Cattle. Genes 2021, 12, 1163. [Google Scholar] [CrossRef]
- May, K.; Scheper, C.; Brügemann, K.; Yin, T.; Strube, C.; Korkuć, P.; Brockmann, G.A.; König, S. Genome-Wide Associations and Functional Gene Analyses for Endoparasite Resistance in an Endangered Population of Native German Black Pied Cattle. BMC Genom. 2019, 20, 277. [Google Scholar] [CrossRef]
- Korkuć, P.; Arends, D.; May, K.; König, S.; Brockmann, G.A. Genomic Loci Affecting Milk Production in German Black Pied Cattle (DSN). Front. Genet. 2021, 12, 640039. [Google Scholar] [CrossRef]
- Korkuć, P.; Neumann, G.B.; Hesse, D.; Arends, D.; Reißmann, M.; Rahmatalla, S.; May, K.; Wolf, M.J.; König, S.; Brockmann, G.A. Whole-Genome Sequencing Data Reveal New Loci Affecting Milk Production in German Black Pied Cattle (DSN). Genes 2023, 14, 581. [Google Scholar] [CrossRef]
- Meier, S.; Arends, D.; Korkuć, P.; Neumann, G.B.; Brockmann, G.A. A Genome-Wide Association Study for Clinical Mastitis in the Dual-Purpose German Black Pied Cattle Breed. J. Dairy Sci. 2020, 103, 10289–10298. [Google Scholar] [CrossRef]
- Neumann, G.B.; Korkuć, P.; Arends, D.; Wolf, M.J.; May, K.; Reißmann, M.; Elzaki, S.; König, S.; Brockmann, G.A. Design and Performance of a Bovine 200 k SNP Chip Developed for Endangered German Black Pied Cattle (DSN). BMC Genom. 2021, 22, 905. [Google Scholar] [CrossRef]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Zimin, A.; Dreischer, C.; Schultheiss, S.; Hall, R.; Schroeder, S.G.; et al. Modernizing the Bovine Reference Genome Assembly. In Proceedings of the 11th World Congress on Genetics Applied to Livestock Production, Auckland, New Zealand, 11–16 February 2018. [Google Scholar]
- Christensen, R.H.B. Ordinal—Regression Models for Ordinal Data. R Package Version 2023.12-4.1, 2023. Available online: https://cran.r-project.org/web/packages/ordinal/index.html (accessed on 27 February 2025).
- Venables, W.N.; Ripley, B.D. Modern Applied Statistics with S, 4th ed.; Springer: New York, NY, USA, 2002; ISBN 0-387-95457-0. [Google Scholar]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.J.; Amode, M.R.; Aneja, A.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Becker, A.; Bennett, R.; Berry, A.; Bhai, J.; et al. Ensembl 2023. Nucleic Acids Res. 2023, 51, D933–D941. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. G:Profiler: A Web Server for Functional Enrichment Analysis and Conversions of Gene Lists (2019 Update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Hu, Z.L.; Park, C.A.; Reecy, J.M. A Combinatorial Approach Implementing New Database Structures to Facilitate Practical Data Curation Management of QTL, Association, Correlation and Heritability Data on Trait Variants. Database 2023, 2023, 24. [Google Scholar] [CrossRef]
- Fantini, D. EasyPubMed: Search and Retrieve Scientific Publication Records from PubMed. R Package Version 2.13, 2019. Available online: https://10.32614/CRAN.package.easyPubMed (accessed on 27 February 2025).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Wickham, H. Reshaping Data with the Reshape Package. J. Stat. Softw. 2007, 21, 1–20. [Google Scholar] [CrossRef]
- Kassambara, A. ggpubr: “ggplot2” Based Publication Ready Plots. R Package Version 0.6.0, 2020. Available online: https://10.32614/CRAN.package.ggpubr (accessed on 27 February 2025).
- Loker, S.; Bastin, C.; Miglior, F.; Sewalem, A.; Schaeffer, L.R.; Jamrozik, J.; Ali, A.; Osborne, V. Genetic and Environmental Relationships between Body Condition Score and Milk Production Traits in Canadian Holsteins. J. Dairy Sci. 2012, 95, 410–419. [Google Scholar] [CrossRef]
- Khmelnychyii, L.; Khmelnychyii, S.; Samokhina, Y. Correlation between Descriptive and Group Type Traits in the System of Cow’s Linear Classification of Ukrainian Brown Dairy Breed. Open Agric. 2023, 8, 20220180. [Google Scholar] [CrossRef]
- Marete, A.G.; Guldbrandtsen, B.; Lund, M.S.; Fritz, S.; Sahana, G.; Boichard, D. A Meta-Analysis Including Pre-Selected Sequence Variants Associated with Seven Traits in Three French Dairy Cattle Populations. Front. Genet. 2018, 9, 399480. [Google Scholar] [CrossRef]
- Bouwman, A.C.; Daetwyler, H.D.; Chamberlain, A.J.; Ponce, C.H.; Sargolzaei, M.; Schenkel, F.S.; Sahana, G.; Govignon-Gion, A.; Boitard, S.; Dolezal, M.; et al. Meta-Analysis of Genome-Wide Association Studies for Cattle Stature Identifies Common Genes That Regulate Body Size in Mammals. Nat. Genet. 2018, 50, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.B.; Wiggans, G.R.; Ma, L.; Sonstegard, T.S.; Lawlor, T.J.; Crooker, B.A.; Van Tassell, C.P.; Yang, J.; Wang, S.; Matukumalli, L.K.; et al. Genome-Wide Association Analysis of Thirty One Production, Health, Reproduction and Body Conformation Traits in Contemporary U.S. Holstein Cows. BMC Genom. 2011, 12, 408. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Zinder, M.; Asher, A.; Lipkin, E.; Feingersch, R.; Agmon, R.; Karasik, D.; Brosh, A.; Shabtay, A. FABP4 Is a Leading Candidate Gene Associated with Residual Feed Intake in Growing Holstein Calves. Physiol. Genom. 2016, 48, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Snelling, W.M.; Allan, M.F.; Keele, J.W.; Kuehn, L.A.; McDaneld, T.; Smith, T.P.L.; Sonstegard, T.S.; Thallman, R.M.; Bennett, G.L. Genome-Wide Association Study of Growth in Crossbred Beef Cattle. J. Anim. Sci. 2010, 88, 837–848. [Google Scholar] [CrossRef]
- Lu, D.; Miller, S.; Sargolzaei, M.; Kelly, M.; Vander Voort, G.; Caldwell, T.; Wang, Z.; Plastow, G.; Moore, S. Genome-Wide Association Analyses for Growth and Feed Efficiency Traits in Beef Cattle1. J. Anim. Sci. 2013, 91, 3612–3633. [Google Scholar] [CrossRef]
- Kolbehdari, D.; Wang, Z.; Grant, J.R.R.; Murdoch, B.; Prasad, A.; Xiu, Z.; Marques, E.; Stothard, P.; Moore, S.S.S. A Whole-Genome Scan to Map Quantitative Trait Loci for Conformation and Functional Traits in Canadian Holstein Bulls. J. Dairy Sci. 2008, 91, 2844–2856. [Google Scholar] [CrossRef]
- Igoshin, A.V.; Yudin, N.S.; Romashov, G.A.; Larkin, D.M. A Multibreed Genome-Wide Association Study for Cattle Leukocyte Telomere Length. Genes 2023, 14, 1596. [Google Scholar] [CrossRef]
- Do, D.N.; Bissonnette, N.; Lacasse, P.; Miglior, F.; Sargolzaei, M.; Zhao, X.; Ibeagha-Awemu, E.M. Genome-Wide Association Analysis and Pathways Enrichment for Lactation Persistency in Canadian Holstein Cattle. J. Dairy Sci. 2017, 100, 1955–1970. [Google Scholar] [CrossRef]
- Babiuk, S.; Horseman, B.; Zhang, C.; Bickis, M.; Kusalik, A.; Schook, L.B.; Abrahamsen, M.S.; Pontarollo, R. BoLA Class I Allele Diversity and Polymorphism in a Herd of Cattle. Immunogenetics 2007, 59, 167–176. [Google Scholar] [CrossRef]
- Chen, S.Y.; Oliveira, H.R.; Schenkel, F.S.; Pedrosa, V.B.; Melka, M.G.; Brito, L.F. Using Imputed Whole-Genome Sequence Variants to Uncover Candidate Mutations and Genes Affecting Milking Speed and Temperament in Holstein Cattle. J. Dairy Sci. 2020, 103, 10383–10398. [Google Scholar] [CrossRef]
- Tijjani, A.; Utsunomiya, Y.T.; Ezekwe, A.G.; Nashiru, O.; Hanotte, O. Genome Sequence Analysis Reveals Selection Signatures in Endangered Trypanotolerant West African Muturu Cattle. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Connor, E.E.; Zhou, Y.; Liu, G.E. The Essence of Appetite: Does Olfactory Receptor Variation Play a Role? J. Anim. Sci. 2018, 96, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Jia, Y.; Lee, J.H.; Kim, Y.; Sekharan, S.; Batista, V.S.; Lee, S.J. Activation of OR1A1 Suppresses PPAR-γ Expression by Inducing HES-1 in Cultured Hepatocytes. Int. J. Biochem. Cell Biol. 2015, 64, 75–80. [Google Scholar] [CrossRef]
- An, S.S.; Liggett, S.B. Taste and Smell GPCRs in the Lung: Evidence for a Previously Unrecognized Widespread Chemosensory System. Cell. Signal. 2018, 41, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.; Bahk, Y.Y.; Lee, N.; Jae, Y.; Cho, Y.H.; Ku, C.R.; Byun, Y.; Lee, E.J.; Kim, M.S.; Koo, J. Olfactory Receptor Olfr544 Responding to Azelaic Acid Regulates Glucagon Secretion in α-Cells of Mouse Pancreatic Islets. Biochem. Biophys. Res. Commun. 2015, 460, 616–621. [Google Scholar] [CrossRef]
- Krause, F.; Mohebian, K.; Delpero, M.; Hesse, D.; Kühn, R.; Arends, D.; Brockmann, G.A. A Deletion Containing a CTCF-Element in Intron 8 of the Bbs7 Gene Is Partially Responsible for Juvenile Obesity in the Berlin Fat Mouse. Mamm. Genome 2022, 33, 465–470. [Google Scholar] [CrossRef]
- Mattick, J.S.; Makunin, I.V. Non-Coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Description | Unit, Range | Min | Mean | Sd | Max |
|---|---|---|---|---|---|---|
| COMDAIRY STRENGTH | Composite score based on linear trait below | points, 65–88 | 70 | 79.1 | 3.0 | 86 |
| LINDAIRY_CHARACTER | Rip angle and openness, and bone flatness (angularity) | scale, 1–9 | 1 | 3.4 | 1.3 | 7 |
| COMBODY CONFORMATION | Composite score based on linear traits below | points, 65–88 | 69 | 79.3 | 3.4 | 87 |
| LINBCS | Area between ischial tuberosities and lumbar spine | scale, 1–9 | 3 | 6.1 | 1.1 | 9 |
| LINBODY_DEPTH | Distance at last rib from spine (top) to barrel (bottom) | scale, 1–9 | 2 | 5.4 | 1.3 | 9 |
| LINCHEST_WIDTH | Width between the top of the front legs | scale, 1–9 | 3 | 6.3 | 1.1 | 9 |
| LINRUMP_ANGLE | Rump structure from hips to pins | scale, 1–9 | 2 | 4.8 | 0.9 | 7 |
| LINRUMP_WIDTH | Distance between pin bones (outer points) | scale, 1–9 | 3 | 5.7 | 1.2 | 9 |
| LINSTATURE | Distance from spine (top between hips) to ground | cm, - | 128 | 136.8 | 2.9 | 145 |
| COMLEGS&FEET | Composite score based on linear traits below | points, 65–88 | 73 | 82.0 | 2.8 | 87 |
| LINFOOT_ANGLE | Angle at the front of the rear right hoof | scale, 1–9 | 3 | 5.1 | 1.0 | 8 |
| LINHOCK_QUALITY | Thickness of the ankle joint | scale, 1–9 | 2 | 6.0 | 1.4 | 9 |
| LINLOCOMOTION | Step length and straightness (movement harmony) | scale, 1–9 | 1 | 5.9 | 1.7 | 9 |
| LINREAR_LEG_REAR | Direction of feet (viewed from rear) | scale, 1–9 | 2 | 5.9 | 1.5 | 9 |
| LINREAR_LEG_SIDE | Angle at the front of the hock | scale, 1–9 | 3 | 5.3 | 0.9 | 8 |
| COMMAMMARY | Composite score based on linear traits below | points, 65–88 | 68 | 78.3 | 3.5 | 87 |
| LINCENTRAL_LIGAMENT | Depth of the cleft at the base of the rear udder | scale, 1–9 | 1 | 4.3 | 1.4 | 8 |
| LINFORE_UDDER | Fore udder attachment strength to abdominal wall | scale, 1–9 | 2 | 5.3 | 1.3 | 9 |
| LINFRONT_TEAT | Front teat position (relative to udder center) | scale, 1–9 | 1 | 4.0 | 1.2 | 7 |
| LINREAR_TEAT | Rear teat position (relative to udder center) | scale, 1–9 | 2 | 5.0 | 1.1 | 8 |
| LINREAR_UDDER | Distance from vulva (bottom) to udder relative to height | scale, 1–9 | 1 | 4.3 | 1.4 | 8 |
| LINTEAT_LENGTH | Length of the front teat | scale, 1–9 | 2 | 4.9 | 1.0 | 7 |
| LINUDDER_DEPTH | Distance from udder (lowest part) to the hock | scale, 1–9 | 1 | 4.4 | 1.2 | 8 |
| COMDAIRY STRENGTH | COMBODY CONFORMATION | COMLEGS&FEET | COMMAMMARY | |
|---|---|---|---|---|
| LINDAIRY_CHARACTER | 0.54 | −0.06 | 0.10 | 0.05 |
| LINBCS | −0.27 | 0.34 | 0.05 | 0.01 |
| LINBODY_DEPTH | 0.20 | 0.66 | 0.03 | 0.07 |
| LINCHEST_WIDTH | −0.05 | 0.53 | 0.01 | 0.05 |
| LINRUMP_ANGLE | −0.10 | −0.06 | −0.04 | −0.08 |
| LINRUMP_WIDTH | 0.13 | 0.52 | 0.05 | 0.05 |
| LINSTATURE | 0.34 | 0.60 | 0.02 | 0.05 |
| LINFOOT_ANGLE | 0.08 | 0.11 | 0.35 | 0.10 |
| LINHOCK_QUALITY | 0.13 | −0.02 | 0.43 | 0.12 |
| LINLOCOMOTION | 0.12 | 0.09 | 0.83 | 0.18 |
| LINREAR_LEG_REAR | 0.15 | 0.11 | 0.61 | 0.17 |
| LINREAR_LEG_SIDE | −0.02 | −0.10 | −0.30 | −0.06 |
| LINCENT._LIGAMENT | 0.12 | 0.01 | 0.09 | 0.36 |
| LINFORE_UDDER | 0.07 | 0.12 | 0.11 | 0.53 |
| LINFRONT_TEAT | 0.11 | 0.11 | 0.15 | 0.47 |
| LINREAR_TEAT | 0.11 | 0.13 | 0.07 | 0.30 |
| LINREAR_UDDER | 0.20 | 0.06 | 0.25 | 0.54 |
| LINTEAT_LENGTH | −0.01 | 0.04 | 0.01 | −0.01 |
| LINUDDER_DEPTH | 0.12 | 0.09 | 0.03 | 0.46 |
| Trait/Score | Chr | Position | rs-ID | MA | MAF | βMA | SE(βMA) | −log10(p) |
|---|---|---|---|---|---|---|---|---|
| COMDAIRY STRENGTH | ||||||||
| LINDAIRY_CHARACTER | 20 | 21,627,470 | rs381893684 | G | 0.50 | 0.43 | 0.07 | 7.23 |
| COMDAIRY STRENGTH | X | 11,548,482 | - | C | 0.30 | −0.86 | 0.12 | 7.26 |
| COMBODY CONFORMATION | ||||||||
| LINBCS | 2 | 109,169,387 | rs378978517 | C | 0.16 | −0.55 | 0.09 | 7.01 |
| LINRUMP_ANGLE | 7 | 98,433,366 | rs110898200 | A | 0.44 | −0.46 | 0.07 | 6.92 |
| LINRUMP_WIDTH | 8 | 15,021,228 | rs109990310 | G | 0.18 | −0.58 | 0.09 | 6.93 |
| LINBCS | 10 | 90,095,068 | rs385227612 | ATG | 0.12 | 0.67 | 0.10 | 6.77 |
| LINRUMP_WIDTH | 21 | 17,993,077 | rs210390204 | T | 0.19 | −0.58 | 0.08 | 8.27 |
| COMBODY CONFORMATION | 21 | 37,111,947 | rs132717442 | A | 0.15 | −1.09 | 0.15 | 7.31 |
| LINBCS | 24 | 20,358,408 | rs134912642 | A | 0.40 | 0.44 | 0.07 | 7.49 |
| LINRUMP_ANGLE | 26 | 21,470,978 | rs110956215 | A | 0.21 | −0.55 | 0.08 | 7.24 |
| COMLEGS&FEET | ||||||||
| COMLEGS&FEET | 6 | 105,886,046 | rs133849982 | C | 0.19 | −0.65 | 0.12 | 7.34 |
| LINLOCOMOTION | 12 | 3,371,041 | rs42662438 | G | 0.22 | 0.41 | 0.08 | 7.25 |
| LINREAR_LEG_SIDE | 13 | 17,654,151 | - | C | 0.08 | 0.77 | 0.13 | 6.92 |
| LINREAR_LEG_SIDE | 19 | 22,241,461 | rs379037369 | C | 0.44 | 0.41 | 0.07 | 6.98 |
| LINREAR_LEG_SIDE | 20 | 29,327,008 | - | G | 0.38 | 0.44 | 0.07 | 7.29 |
| LINHOCK_QUALITY | 23 | 28,684,391 | rs800639948 | GA | 0.15 | −0.52 | 0.09 | 6.90 |
| COMLEGS&FEET | 24 | 10,452,692 | rs110128208 | T | 0.09 | −0.92 | 0.17 | 6.78 |
| COMLEGS&FEET | 26 | 5,538,549 | rs135255414 | T | 0.17 | −0.62 | 0.12 | 6.80 |
| COMMAMMARY | ||||||||
| LINFORE_UDDER | 4 | 21,420,050 | rs134371955 | G | 0.50 | −0.37 | 0.06 | 7.11 |
| LINREAR_UDDER | 6 | 15,034,445 | - | T | 0.05 | −0.89 | 0.15 | 7.35 |
| LINCENTRAL_LIGAMENT | 15 | 50,014,862 | - | G | 0.38 | 0.38 | 0.06 | 7.21 |
| LINFORE_UDDER | 17 | 14,049,897 | rs379498892 | A | 0.34 | −0.39 | 0.07 | 7.25 |
| LINCENTRAL_LIGAMENT | 18 | 16,330,666 | rs110023445 | G | 0.38 | −0.38 | 0.06 | 7.06 |
| LINTEAT_LENGTH | 22 | 17,411,573 | rs378674848 | T | 0.10 | −0.78 | 0.11 | 7.91 |
| Chr | Position | QTL Start | QTL End | Positional Candidate Genes (Number of Genes) |
| COMDAIRY STRENGTH | ||||
| 20 | 21,627,470 | 21,477,470 | 21,777,470 | - (0) |
| X | 11,548,482 | 11,398,482 | 11,698,482 | - (0) |
| COMBODY CONFORMATION | ||||
| 2 | 109,169,387 | 109,019,387 | 109,319,387 | - (0) |
| 7 | 98,433,366 | 98,283,366 | 98,583,366 | - (0) |
| 8 | 15,021,228 | 14,871,228 | 15,171,228 | - (0) |
| 10 | 90,095,068 | 89,945,068 | 90,245,068 | NRXN3 (1) |
| 21 | 17,993,077 | 17,843,077 | 18,143,077 | - (0) |
| 21 | 37,111,947 | 36,961,947 | 37,261,947 | - (0) |
| 24 | 20,358,408 | 20,208,408 | 20,508,408 | FHOD3, TPGS2, ENSBTAG00000048612 (3) |
| 26 | 21,470,978 | 21,320,978 | 21,620,978 | SEC31B, NDUFB8, HIF1AN, WNT8B, PAX2 (5) |
| COMLEGS&FEET | ||||
| 6 | 105,886,046 | 105,736,046 | 106,036,046 | - (0) |
| 12 | 3,371,041 | 3,221,041 | 3,521,041 | ENSBTAG00000051623 (1) |
| 13 | 17,654,151 | 17,504,151 | 17,804,151 | FBH1, IL15RA, ANKRD26, YME1L1, MASTL, ACBD5, ENSBTAG00000053950 (7) |
| 19 | 22,241,461 | 22,091,461 | 22,391,461 | RFLNB, GEMIN4, VPS53, RPH3AL, C19H17orf97, TLCD3A, ENSBTAG00000054644 (7) |
| 20 | 29,327,008 | 29,177,008 | 29,477,008 | HCN1 (1) |
| 23 | 28,684,391 | 28,534,391 | 28,834,391 | BOLA, TRIM10, TRIM15, BOLA-NC1, JSP.1, TRIM26, TRIM40, ENSBTAG00000037421 (8) |
| 24 | 10,452,692 | 10,302,692 | 10,602,692 | - (0) |
| 26 | 5,538,549 | 5,388,549 | 5,688,549 | PCDH15 (1) |
| COMMAMMARY | ||||
| 4 | 21,420,050 | 21,270,050 | 22,207,653 | ETV1 (1) |
| 6 | 15,034,445 | 14,884,445 | 15,184,445 | ENPEP, ENSBTAG00000054468 (2) |
| 15 | 50,014,862 | 49,373,191 | 50,164,862 | OR51AB4, OR52AB4, OR52AE11, OR51G4, OR52J3D, ENSBTAG00000038578, OR51AG1, ENSBTAG00000039185, OR51P1, OR51V22, OR52Z12, OR52AB9, OR52E58, OR52S27, OR52Z19, OR52AD1, OR52A24, OR51L15, OR51G2, OR51L1B, ENSBTAG00000049986, ENSBTAG00000050365, OR51V1B, ENSBTAG00000051323, OR52J1, OR52S6, OR52E1, OR51S1B, OR51P1B, OR51L22, OR51L1C, OR52S20, OR51A8, OR51H5 (34) |
| 17 | 14,049,897 | 13,899,897 | 14,281,310 | GYPA, GYPB (2) |
| 18 | 16,330,666 | 16,180,666 | 16,480,666 | - (0) |
| 22 | 17,411,573 | 17,261,573 | 17,561,573 | SRGAP3 (1) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandel, A.; Reißmann, M.; Brockmann, G.A.; Korkuć, P. Whole-Genome Insights into the Genetic Basis of Conformation Traits in German Black Pied (DSN) Cattle. Genes 2025, 16, 445. https://doi.org/10.3390/genes16040445
Mandel A, Reißmann M, Brockmann GA, Korkuć P. Whole-Genome Insights into the Genetic Basis of Conformation Traits in German Black Pied (DSN) Cattle. Genes. 2025; 16(4):445. https://doi.org/10.3390/genes16040445
Chicago/Turabian StyleMandel, Amelie, Monika Reißmann, Gudrun A. Brockmann, and Paula Korkuć. 2025. "Whole-Genome Insights into the Genetic Basis of Conformation Traits in German Black Pied (DSN) Cattle" Genes 16, no. 4: 445. https://doi.org/10.3390/genes16040445
APA StyleMandel, A., Reißmann, M., Brockmann, G. A., & Korkuć, P. (2025). Whole-Genome Insights into the Genetic Basis of Conformation Traits in German Black Pied (DSN) Cattle. Genes, 16(4), 445. https://doi.org/10.3390/genes16040445