
Profiling of Known and Novel microRNAs in an Oleaginous Crop Native to the Amazon Basin, Sacha Inchi (Plukenetia volubilis), Through smallRNA-Seq

 , , , and
, , , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cultivation of Sacha Inchi and Sample Preparation
2.2. Extraction of smallRNA and Sequencing of microRNA
2.3. Pre-Processing of Sequencing Data
2.4. Identification of Known and Novel microRNAs
2.5. Differential Expression Analysis of miRNAs Between Organs
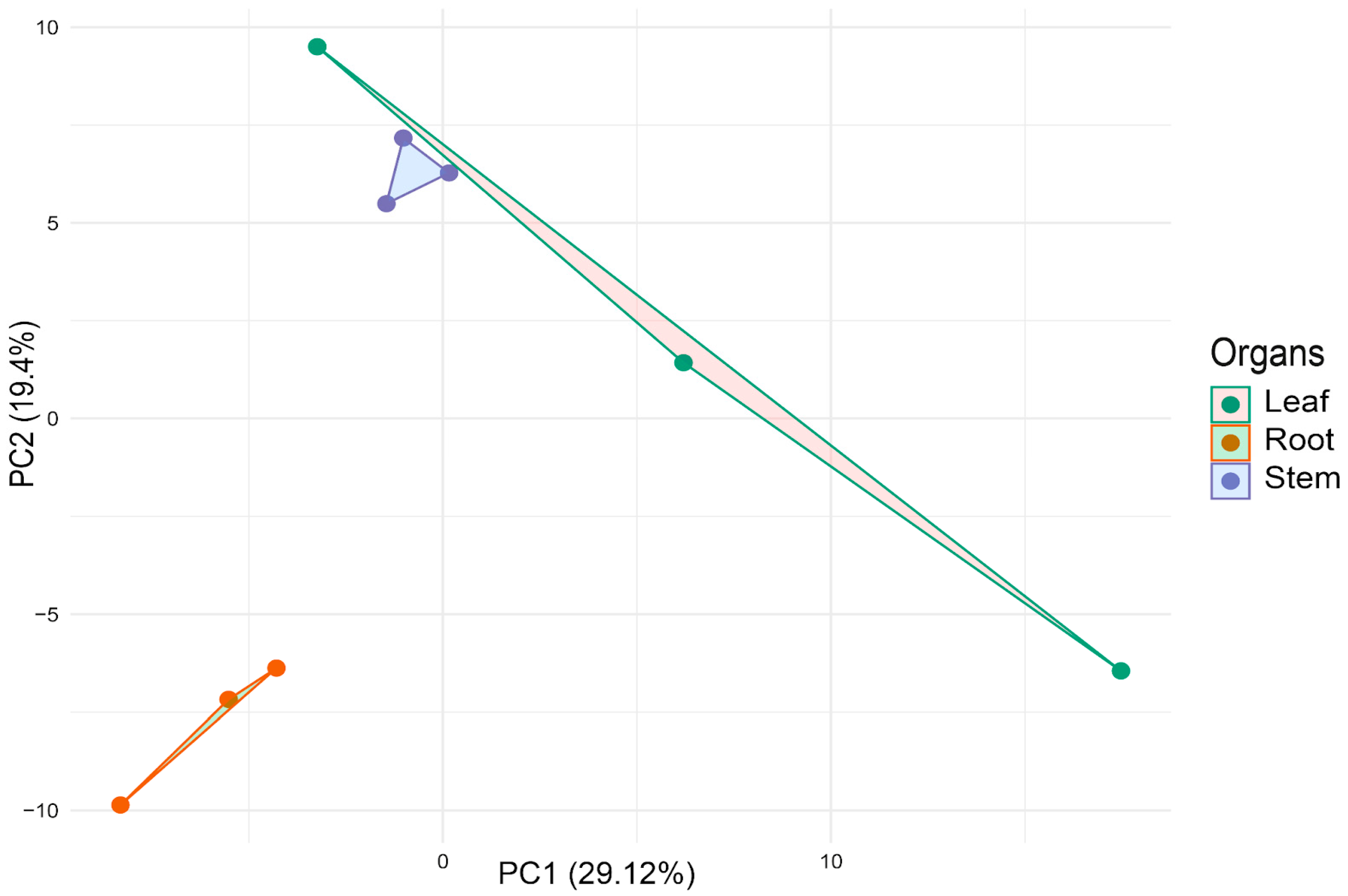
2.6. Multivariate Analysis of miRNA Expression Profiles
2.7. Prediction of Target Genes
2.8. Functional Enrichment Analysis of Target Genes: GO and KEGG Pathways
3. Results
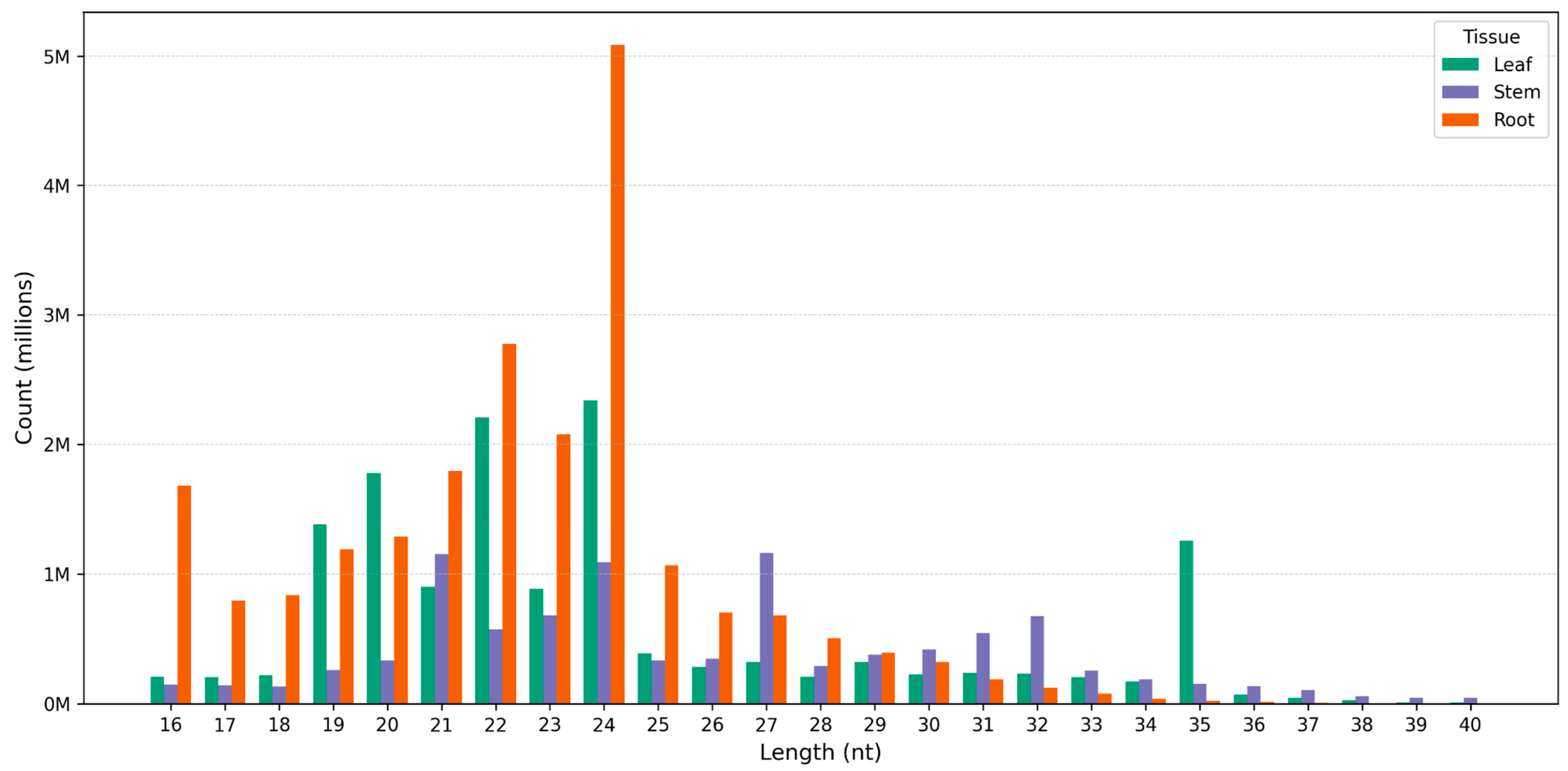
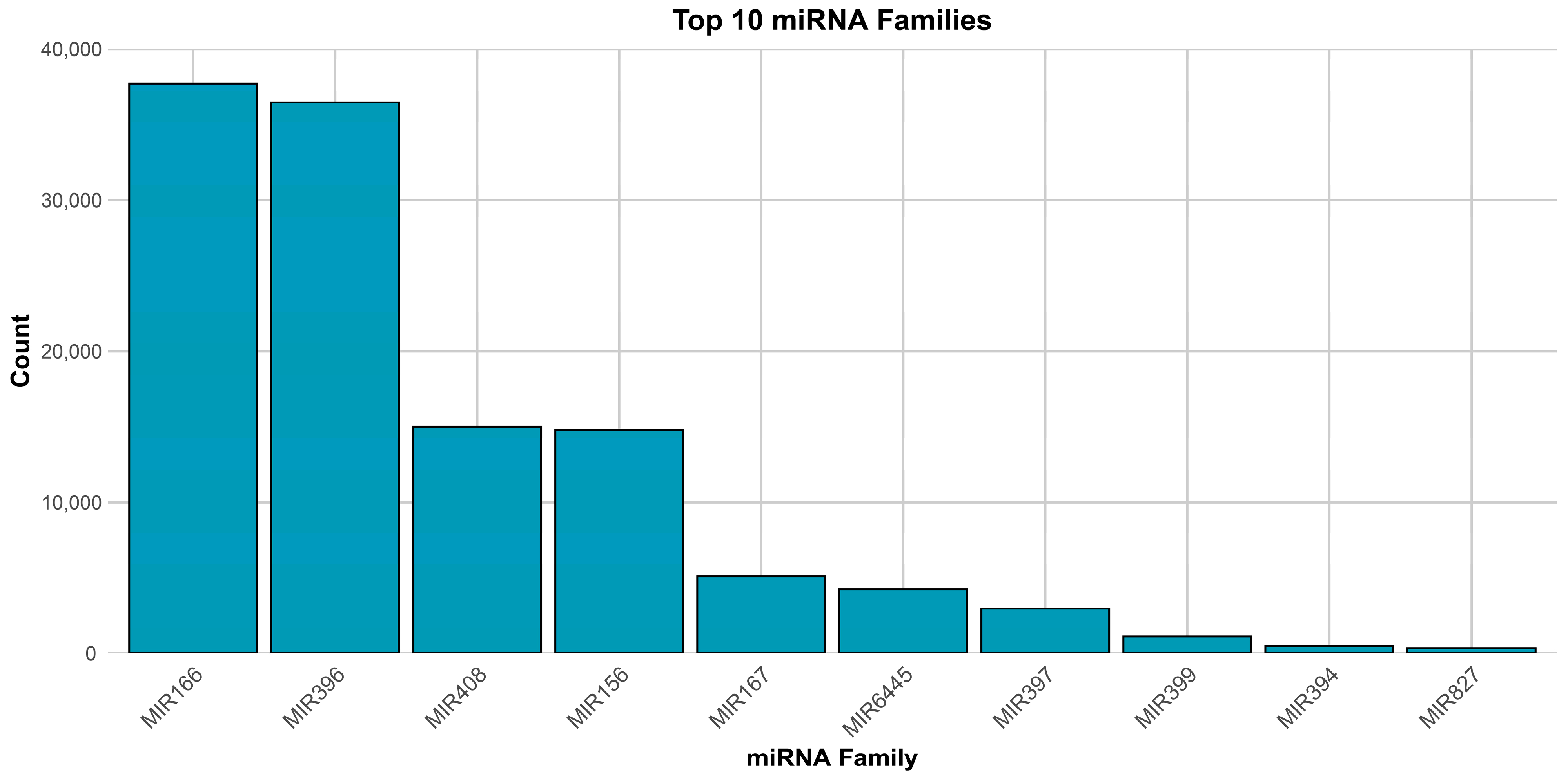
3.1. Profile of Known and Novel microRNAs
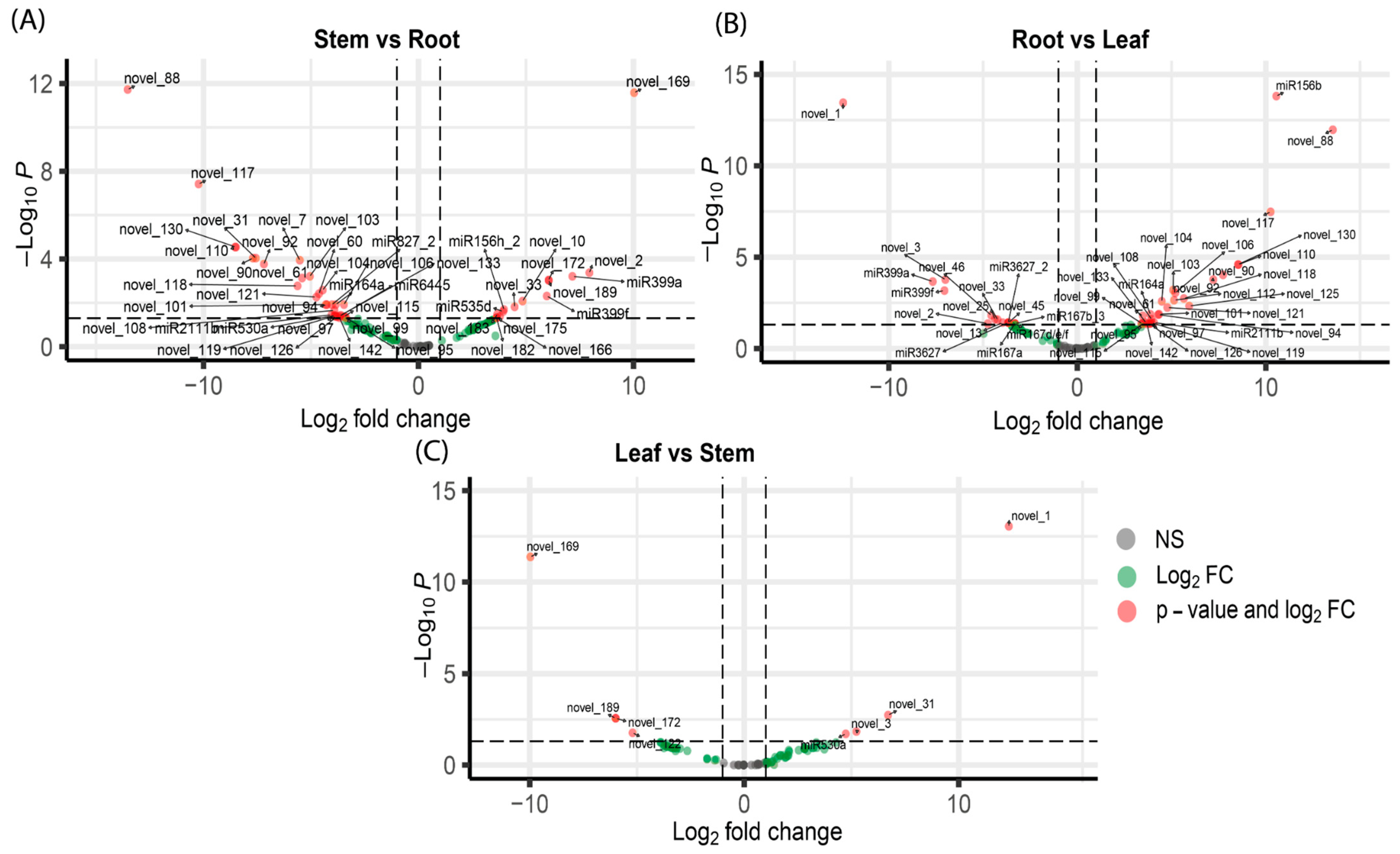
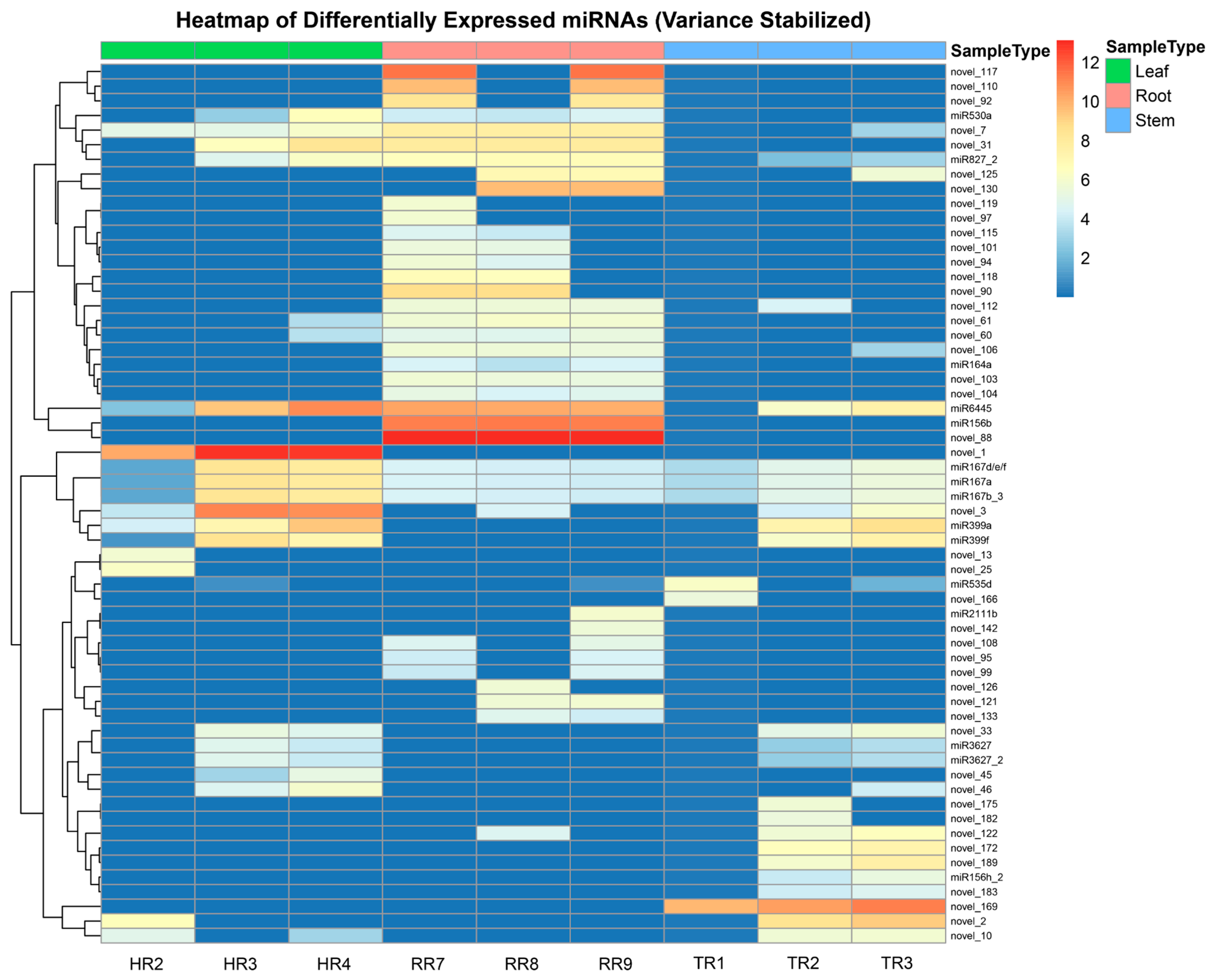
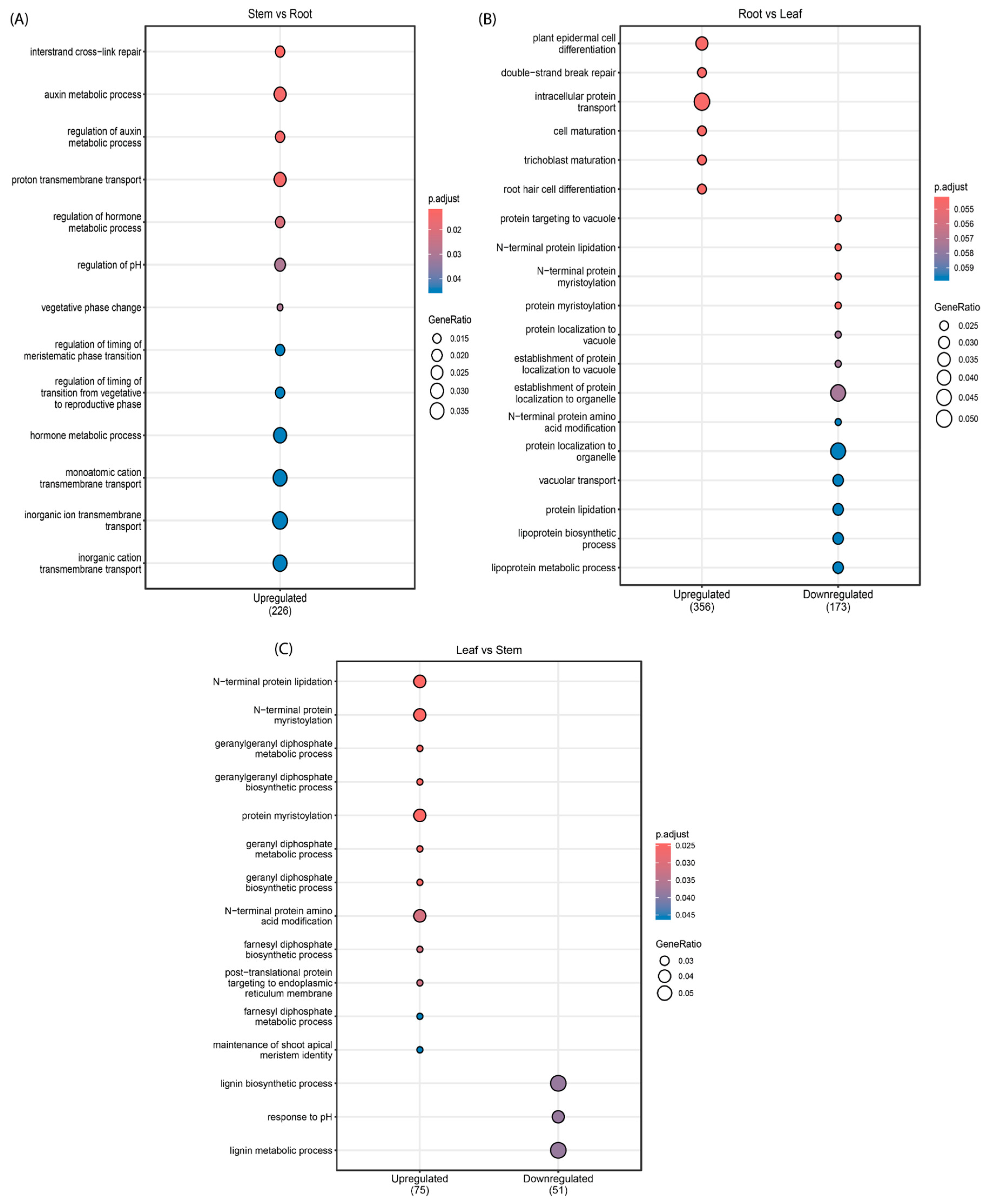
3.2. Differential Expression Analysis of miRNAs and Their Target Genes
3.3. Potential Targets of Novel miRNAs in Sacha Inchi
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hu, X.-D.; Pan, B.-Z.; Fu, Q.; Niu, L.; Chen, M.-S.; Xu, Z.-F. De Novo Transcriptome Assembly of the Eight Major Organs of Sacha Inchi (Plukenetia volubilis) and the Identification of Genes Involved in α-Linolenic Acid Metabolism. BMC Genom. 2018, 19, 380. [Google Scholar] [CrossRef]
- Yamuangmorn, S.; Laororng, K.; Saenchai, C.; Lordkaew, S.; Dell, B.; Prom-u-Thai, C. Boron Requirement for Vegetative Growth of Sacha Inchi (Plukentia volubilis L.). J. Plant Nutr. 2022, 45, 845–853. [Google Scholar] [CrossRef]
- Wiriya, J.; Rangjaroen, C.; Teaumroong, N.; Sungthong, R.; Lumyong, S. Rhizobacteria and Arbuscular Mycorrhizal Fungi of Oil Crops (Physic Nut and Sacha Inchi): A Cultivable-Based Assessment for Abundance, Diversity, and Plant Growth-Promoting Potentials. Plants 2020, 9, 1773. [Google Scholar] [CrossRef]
- Lakhwani, D.; Sanchita; Pandey, A.; Sharma, D.; Asif, M.H.; Trivedi, P.K. Novel microRNAs Regulating Ripening-Associated Processes in Banana Fruit. Plant Growth Regul. 2020, 90, 223–235. [Google Scholar] [CrossRef]
- Gao, Z.; Nie, J.; Wang, H. MicroRNA Biogenesis in Plant. Plant Growth Regul. 2021, 93, 1–12. [Google Scholar] [CrossRef]
- Yu, Y.; Jia, T.; Chen, X. The ‘How’ and ‘Where’ of Plant microRNAs. New Phytol. 2017, 216, 1002–1017. [Google Scholar] [CrossRef]
- Baroin-Tourancheau, A.; Benigni, X.; Doubi-Kadmiri, S.; Taouis, M.; Amar, L. Lessons from microRNA Sequencing Using Illumina Technology. Adv. Biosci. Biotechnol. 2016, 7, 319–328. [Google Scholar] [CrossRef]
- Buermans, H.P.; Ariyurek, Y.; van Ommen, G.; den Dunnen, J.T.; ’t Hoen, P.A. New Methods for next Generation Sequencing Based microRNA Expression Profiling. BMC Genom. 2010, 11, 716. [Google Scholar] [CrossRef]
- Benesova, S.; Kubista, M.; Valihrach, L. Small RNA-Sequencing: Approaches and Considerations for miRNA Analysis. Diagnostics 2021, 11, 964. [Google Scholar] [CrossRef]
- Liu, G.; Wu, Z.; Peng, Y.; Shang, X.; Gao, L. Integrated Transcriptome and Proteome Analysis Provides Insight into the Ribosome Inactivating Proteins in Plukenetia volubilis Seeds. Int. J. Mol. Sci. 2022, 23, 9562. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Niu, Q.; Dai, H.; Tian, X.; Ma, J.; Pritchard, H.W.; Lin, L.; Yang, X. The Transcription Factor MYB1 Activates DGAT2 Transcription to Promote Triacylglycerol Accumulation in Sacha Inchi (Plukenetia volubilis L.) Leaves under Heat Stress. Plant Physiol. Biochem. 2024, 208, 108517. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Niu, L.; Chen, M.-S.; Tao, Y.-B.; Wang, X.; He, H.; Pan, B.-Z.; Xu, Z.-F. De Novo Transcriptome Assembly and Comparative Analysis between Male and Benzyladenine-Induced Female Inflorescence Buds of Plukenetia volubilis. J. Plant Physiol. 2018, 221, 107–118. [Google Scholar] [CrossRef]
- Babraham Bioinformatics—FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 28 November 2024).
- Krueger, F. FelixKrueger/TrimGalore 2024. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 28 November 2024).
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Ontiveros-Palacios, N.; Cooke, E.; Nawrocki, E.P.; Triebel, S.; Marz, M.; Rivas, E.; Griffiths-Jones, S.; Petrov, A.I.; Bateman, A.; Sweeney, B. Rfam 15: RNA Families Database in 2025. Nucleic Acids Res. 2024, 53, D258–D267. [Google Scholar] [CrossRef] [PubMed]
- RNAcentral Consortium. RNAcentral 2021: Secondary Structure Integration, Improved Sequence Search and New Member Databases. Nucleic Acids Res. 2021, 49, D212–D220. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Inokuchi, H.; Yamada, Y.; Muto, A.; Iwasaki, Y.; Ikemura, T. tRNADB-CE: tRNA Gene Database Well-Timed in the Era of Big Sequence Data. Front. Genet. 2014, 5, 114. [Google Scholar] [CrossRef]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- Luo, X.; Chen, S.; Zhang, Y. PlantRep: A Database of Plant Repetitive Elements. Plant Cell Rep. 2022, 41, 1163–1166. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and Memory-Efficient Alignment of Short DNA Sequences to the Human Genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Lu, H.; Xue, F.; Ma, L. Identifying High Confidence microRNAs in the Developing Seeds of Jatropha Curcas. Sci. Rep. 2019, 9, 4510. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, L. miRDeep-P: A Computational Tool for Analyzing the microRNA Transcriptome in Plants. Bioinformatics 2011, 27, 2614–2615. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating High Confidence microRNAs Using Deep Sequencing Data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Hofacker, I.L. Vienna RNA Secondary Structure Server. Nucleic Acids Res. 2003, 31, 3429–3431. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Prediction of RNA Secondary Structure by Energy Minimization. In Computer Analysis of Sequence Data: Part II; Griffin, A.M., Griffin, H.G., Eds.; Springer: Totowa, NJ, USA, 1994; pp. 267–294. ISBN 978-1-59259-512-9. [Google Scholar]
- Zuker, M.; Stiegler, P. Optimal Computer Folding of Large RNA Sequences Using Thermodynamics and Auxiliary Information. Nucleic Acids Res. 1981, 9, 133–148. [Google Scholar] [CrossRef]
- Kumar, D.; Kumar, S.; Ayachit, G.; Bhairappanavar, S.B.; Ansari, A.; Sharma, P.; Soni, S.; Das, J. Cross-Kingdom Regulation of Putative miRNAs Derived from Happy Tree in Cancer Pathway: A Systems Biology Approach. Int. J. Mol. Sci. 2017, 18, 1191. [Google Scholar] [CrossRef] [PubMed]
- Devi, K.J.; Chakraborty, S.; Deb, B.; Rajwanshi, R. Computational Identification and Functional Annotation of microRNAs and Their Targets from Expressed Sequence Tags (ESTs) and Genome Survey Sequences (GSSs) of Coffee (Coffea arabica L.). Plant Gene 2016, 6, 30–42. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Mullan, K.A.; Bramberger, L.M.; Munday, P.R.; Goncalves, G.; Revote, J.; Mifsud, N.A.; Illing, P.T.; Anderson, A.; Kwan, P.; Purcell, A.W.; et al. ggVolcanoR: A Shiny App for Customizable Visualization of Differential Expression Datasets. Comput. Struct. Biotechnol. J. 2021, 19, 5735–5740. [Google Scholar] [CrossRef]
- Dixon, P. VEGAN, a Package of R Functions for Community Ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Wickham, H. Data Analysis. In ggplot2: Elegant Graphics for Data Analysis; Wickham, H., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 189–201. ISBN 978-3-319-24277-4. [Google Scholar]
- Dai, X.; Zhuang, Z.; Zhao, P.X. psRNATarget: A Plant Small RNA Target Analysis Server (2017 Release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A. topGO. Bioconductor 2019. Available online: https://bioconductor.org/packages/release/bioc/html/topGO.html (accessed on 27 November 2024).
- Carlson, M. Genome Wide Annotation for Arabidopsis. Available online: http://bioconductor.org/packages/org.At.tair.db/ (accessed on 27 November 2024).
- Xu, L.; Hu, Y.; Cao, Y.; Li, J.; Ma, L.; Li, Y.; Qi, Y. An Expression Atlas of miRNAs in Arabidopsis Thaliana. Sci. China Life Sci. 2018, 61, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.; De Meyer, T.; Vandepoele, K.; Kyndt, T. Spatiotemporal Expression Profile of Novel and Known Small RNAs throughout Rice Plant Development Focussing on Seed Tissues. BMC Genom. 2022, 23, 44. [Google Scholar] [CrossRef] [PubMed]
- Grabowska, A.; Bhat, S.S.; Smoczynska, A.; Bielewicz, D.; Jarmolowski, A.; Kulinska, Z.S. Regulation of Plant microRNA Biogenesis. In Plant microRNAs: Shaping Development and Environmental Responses; Miguel, C., Dalmay, T., Chaves, I., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 3–24. ISBN 978-3-030-35772-6. [Google Scholar]
- Pholo, M.; Coetzee, B.; Maree, H.J.; Young, P.R.; Lloyd, J.R.; Kossmann, J.; Hills, P.N. Cell Division and Turgor Mediate Enhanced Plant Growth in Arabidopsis Plants Treated with the Bacterial Signalling Molecule Lumichrome. Planta 2018, 248, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Nidhi, S.; Preciado, J.; Tie, L. Knox Homologs Shoot Meristemless (STM) and KNAT6 Are Epistatic to CLAVATA3 (CLV3) during Shoot Meristem Development in Arabidopsis Thaliana. Mol. Biol. Rep. 2021, 48, 6291–6302. [Google Scholar] [CrossRef]
- Wu, Z.; Liang, J.; Wang, C.; Zhao, X.; Zhong, X.; Cao, X.; Li, G.; He, J.; Yi, M. Overexpression of Lily HsfA3s in Arabidopsis Confers Increased Thermotolerance and Salt Sensitivity via Alterations in Proline Catabolism. J. Exp. Bot. 2018, 69, 2005–2021. [Google Scholar] [CrossRef] [PubMed]
- Mandal, K. Review of PIP2 in Cellular Signaling, Functions and Diseases. Int. J. Mol. Sci. 2020, 21, 8342. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Q.; Yang, G.; Zhang, C.; Dong, H.; Liu, Y.; Yin, R.; Lin, L. Pivotal Roles of Tomato Photoreceptor SlUVR8 in Seedling Development and UV-B Stress Tolerance. Biochem. Biophys. Res. Commun. 2020, 522, 177–183. [Google Scholar] [CrossRef]
- Matsumura, Y.; Iwakawa, H.; Machida, Y.; Machida, C. Characterization of Genes in the ASYMMETRIC LEAVES2/LATERAL ORGAN BOUNDARIES (AS2/LOB) Family in Arabidopsis Thaliana, and Functional and Molecular Comparisons between AS2 and Other Family Members. Plant J. 2009, 58, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Lei, X.; Lü, J.; Gao, C. Overexpression of the ThTPS Gene Enhanced Salt and Osmotic Stress Tolerance in Tamarix Hispida. J. For. Res. 2022, 33, 299–308. [Google Scholar] [CrossRef]
- Hála, M.; Soukupová, H.; Synek, L.; Žárský, V. Arabidopsis RAB Geranylgeranyl Transferase β-Subunit Mutant Is Constitutively Photomorphogenic, and Has Shoot Growth and Gravitropic Defects. Plant J. 2010, 62, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Yu, N.; Niu, Q.-W.; Ng, K.-H.; Chua, N.-H. The Role of miR156/SPLs Modules in Arabidopsis Lateral Root Development. Plant J. 2015, 83, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Shuai, P.; Su, Y.; Liang, D.; Zhang, Z.; Xia, X.; Yin, W. Identification of phasiRNAs and Their Drought—Responsiveness in Populus Trichocarpa. FEBS Lett. 2016, 590, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Yang, X.; Song, Y.; Du, Q.; Li, Y.; Chen, J.; Zhang, D. Adaptive Evolution and Functional Innovation of Populus-Specific Recently Evolved microRNAs. New Phytol. 2017, 213, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Jardim-Messeder, D.; de Souza-Vieira, Y.; Lavaquial, L.C.; Cassol, D.; Galhego, V.; Bastos, G.A.; Felix-Cordeiro, T.; Corrêa, R.L.; Zámocký, M.; Margis-Pinheiro, M.; et al. Ascorbate-Glutathione Cycle Genes Families in Euphorbiaceae: Characterization and Evolutionary Analysis. Biology 2023, 12, 19. [Google Scholar] [CrossRef]
- Zheng, L.; Nagpal, P.; Villarino, G.; Trinidad, B.; Bird, L.; Huang, Y.; Reed, J.W. miR167 Limits Anther Growth to Potentiate Anther Dehiscence. Development 2019, 146, dev.174375. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Huang, S.; Xie, H. Advances in the Regulation of Plant Development and Stress Response by miR167. Front. Biosci. Landmark 2021, 26, 655–665. [Google Scholar] [CrossRef]
- Pegler, J.L.; Oultram, J.M.J.; Grof, C.P.L.; Eamens, A.L. Molecular Manipulation of the miR399/PHO2 Expression Module Alters the Salt Stress Response of Arabidopsis Thaliana. Plants 2021, 10, 73. [Google Scholar] [CrossRef]
- Liu, J.; Ren, Y.; Sun, Y.; Yin, Y.; Han, B.; Zhang, L.; Song, Y.; Zhang, Z.; Xu, Y.; Fan, D.; et al. Identification and Analysis of the MIR399 Gene Family in Grapevine Reveal Their Potential Functions in Abiotic Stress. Int. J. Mol. Sci. 2024, 25, 2979. [Google Scholar] [CrossRef]
- Sun, Y.; Xiong, X.; Wang, Q.; Zhu, L.; Wang, L.; He, Y.; Zeng, H. Integrated Analysis of Small RNA, Transcriptome, and Degradome Sequencing Reveals the MiR156, MiR5488 and MiR399 Are Involved in the Regulation of Male Sterility in PTGMS Rice. Int. J. Mol. Sci. 2021, 22, 2260. [Google Scholar] [CrossRef] [PubMed]
- Yaşar, S.; Pulat, E.; Çakır, Ö. Effects of Nitrogen Deficiency and Drought Stresses on miRNA Expressions in Arabidopsis Thaliana. Plant Cell. Tissue Organ Cult. PCTOC 2024, 157, 42. [Google Scholar] [CrossRef]
- Zhao, Y.; Lin, S.; Qiu, Z.; Cao, D.; Wen, J.; Deng, X.; Wang, X.; Lin, J.; Li, X. MicroRNA857 Is Involved in the Regulation of Secondary Growth of Vascular Tissues in Arabidopsis. Plant Physiol. 2015, 169, 2539–2552. [Google Scholar] [CrossRef] [PubMed]
- Milhinhos, A.; Lopes, S.; Miguel, C. microRNA-Mediated Regulation of Plant Vascular Development and Secondary Growth. In Plant microRNAs: Shaping Development and Environmental Responses; Miguel, C., Dalmay, T., Chaves, I., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 143–168. ISBN 978-3-030-35772-6. [Google Scholar]
- Liu, T.-Y.; Lin, W.-Y.; Huang, T.-K.; Chiou, T.-J. MicroRNA-Mediated Surveillance of Phosphate Transporters on the Move. Trends Plant Sci. 2014, 19, 647–655. [Google Scholar] [CrossRef]
- Kant, S.; Peng, M.; Rothstein, S.J. Genetic Regulation by NLA and MicroRNA827 for Maintaining Nitrate-Dependent Phosphate Homeostasis in Arabidopsis. PLoS Genet. 2011, 7, e1002021. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-Y.; Huang, T.-K.; Chiou, T.-J. NITROGEN LIMITATION ADAPTATION, a Target of MicroRNA827, Mediates Degradation of Plasma Membrane–Localized Phosphate Transporters to Maintain Phosphate Homeostasis in Arabidopsis. Plant Cell 2013, 25, 4061–4074. [Google Scholar] [CrossRef]
- Zheng, C.; Ye, M.; Sang, M.; Wu, R. A Regulatory Network for miR156-SPL Module in Arabidopsis Thaliana. Int. J. Mol. Sci. 2019, 20, 6166. [Google Scholar] [CrossRef]
- Chi, Q.; Du, L.; Ma, W.; Niu, R.; Wu, B.; Guo, L.; Ma, M.; Liu, X.; Zhao, H. The miR164-TaNAC14 Module Regulates Root Development and Abiotic-Stress Tolerance in Wheat Seedlings. J. Integr. Agric. 2023, 22, 981–998. [Google Scholar] [CrossRef]
- Cárdenas, F.D.F.R.; Suárez, Y.R.; Rangel, R.M.C.; Garcia, V.L.; Aguilera, K.L.G.; Martínez, N.M.; De Folter, S. Effect of Constitutive miR164 Expression on Plant Morphology and Fruit Development in Arabidopsis and Tomato. Agronomy 2017, 7, 48. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H. The miR156/SPL Module, a Regulatory Hub and Versatile Toolbox, Gears up Crops for Enhanced Agronomic Traits. Mol. Plant 2015, 8, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Zhang, X.; Zhang, H.; Gu, Y.; Huang, X.; Huang, H.; Liu, H.; Zhang, J.; Hu, Y.; Li, Y.; et al. The miR164-Dependent Regulatory Pathway in Developing Maize Seed. Mol. Genet. Genom. 2019, 294, 501–517. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Wang, H.; Tang, X. NAC Transcription Factors in Plant Multiple Abiotic Stress Responses: Progress and Prospects. Front. Plant Sci. 2015, 6, 902. [Google Scholar] [CrossRef]
- Welner, D.H.; Deeba, F.; Lo Leggio, L.; Skriver, K. Chapter 13—NAC Transcription Factors: From Structure to Function in Stress-Associated Networks. In Plant Transcription Factors; Gonzalez, D.H., Ed.; Academic Press: Boston, MA, USA, 2016; pp. 199–212. ISBN 978-0-12-800854-6. [Google Scholar]
- Wang, Y.; Luo, Z.; Zhao, X.; Cao, H.; Wang, L.; Liu, S.; Wang, C.; Liu, M.; Wang, L.; Liu, Z. Superstar microRNA, miR156, Involved in Plant Biological Processes and Stress Response: A Review. Sci. Hortic. 2023, 316, 112010. [Google Scholar] [CrossRef]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.-W.; Weigel, D.; Poethig, R.S. The Sequential Action of miR156 and miR172 Regulates Developmental Timing in Arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Bari, R.; Datt Pant, B.; Stitt, M.; Scheible, W.-R. PHO2, MicroRNA399, and PHR1 Define a Phosphate-Signaling Pathway in Plants. Plant Physiol. 2006, 141, 988–999. [Google Scholar] [CrossRef] [PubMed]
- Scheible, W.-R.; Pant, B.D.; Musialak-Lange, M.; Nuc, P. Nutrient-Responsive Plant microRNAs. In Non Coding RNAs in Plants; Erdmann, V.A., Barciszewski, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 313–337. ISBN 978-3-642-19454-2. [Google Scholar]
- Arnaud, N.; Laufs, P. Plant miRNA Integrated Functions in Development and Reproduction. Front. Plant Physiol. 2023, 1, 1271423. [Google Scholar] [CrossRef]
- Niu, M.-X.; Feng, C.-H.; He, F.; Zhang, H.; Bao, Y.; Liu, S.-J.; Liu, X.; Su, Y.; Liu, C.; Wang, H.-L.; et al. The miR6445- Module Regulates Drought Tolerance by Regulating the Expression of Glutathione S-Transferase U23 and Reactive Oxygen Species Scavenging in Populus. New Phytol. 2024, 242, 2043–2058. [Google Scholar] [CrossRef]
- Yue, E.; Cao, H.; Liu, B. OsmiR535, a Potential Genetic Editing Target for Drought and Salinity Stress Tolerance in Oryza sativa. Plants 2020, 9, 1337. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Wang, X.; Qu, S.; Shen, T.; Li, M.; Zhu, L. The Roles of miR156 in Abiotic and Biotic Stresses in Plants. Plant Physiol. Biochem. 2023, 204, 108150. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Charagh, S.; Karikari, B.; Sharif, R.; Yadav, V.; Mubarik, M.S.; Habib, M.; Zhuang, Y.; Zhang, C.; Chen, H.; et al. miRNAs for Crop Improvement. Plant Physiol. Biochem. 2023, 201, 107857. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.-X.; Feng, Q.; Cao, X.-L.; Zhu, Y.; Wang, H.; Chandran, V.; Fan, J.; Zhao, J.-Q.; Pu, M.; Li, Y.; et al. Osa-miR167d Facilitates Infection of Magnaporthe Oryzae in Rice. J. Integr. Plant Biol. 2020, 62, 702–715. [Google Scholar] [CrossRef]
- Wang, Y.; Li, K.; Chen, L.; Zou, Y.; Liu, H.; Tian, Y.; Li, D.; Wang, R.; Zhao, F.; Ferguson, B.J.; et al. MicroRNA167-Directed Regulation of the Auxin Response Factors GmARF8a and GmARF8b Is Required for Soybean Nodulation and Lateral Root Development. Plant Physiol. 2015, 168, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Ražná, K.; Harenčár, Ľ.; Kučka, M. The Involvement of microRNAs in Plant Lignan Biosynthesis—Current View. Cells 2022, 11, 2151. [Google Scholar] [CrossRef] [PubMed]
- Adjei, M.O.; Zhou, X.; Mao, M.; Rafique, F.; Ma, J. MicroRNAs Roles in Plants Secondary Metabolism. Plant Signal. Behav. 2021, 16, 1915590. [Google Scholar] [CrossRef]
- Okuma, N.; Soyano, T.; Suzaki, T.; Kawaguchi, M. MIR2111-5 Locus and Shoot-Accumulated Mature miR2111 Systemically Enhance Nodulation Depending on HAR1 in Lotus japonicus. Nat. Commun. 2020, 11, 5192. [Google Scholar] [CrossRef] [PubMed]
- Tsikou, D.; Yan, Z.; Holt, D.B.; Abel, N.B.; Reid, D.E.; Madsen, L.H.; Bhasin, H.; Sexauer, M.; Stougaard, J.; Markmann, K. Systemic Control of Legume Susceptibility to Rhizobial Infection by a Mobile microRNA. Science 2018, 362, 233–236. [Google Scholar] [CrossRef]
- Sakuraba, Y.; Yang, M.; Yanagisawa, S. HASTY-Mediated miRNA Dynamics Modulate Nitrogen Starvation-Induced Leaf Senescence in Arabidopsis. Nat. Commun. 2024, 15, 7913. [Google Scholar] [CrossRef]
- Boerjan, W.; Ralph, J.; Baucher, M. Lignin Biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, Y.; Xu, J.; Niu, X.; Qi, J.; Tao, A.; Zhang, L.; Fang, P.; Lin, L.; Su, J. The CCoAOMT1 Gene from Jute (Corchorus capsularis L.) Is Involved in Lignin Biosynthesis in Arabidopsis Thaliana. Gene 2014, 546, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Koshiba, T.; Murakami, S.; Hattori, T.; Mukai, M.; Takahashi, A.; Miyao, A.; Hirochika, H.; Suzuki, S.; Sakamoto, M.; Umezawa, T. CAD2 Deficiency Causes Both Brown Midrib and Gold Hull and Internode Phenotypes in Oryza sativa L. cv. Nipponbare. Plant Biotechnol. 2013, 30, 365–373. [Google Scholar] [CrossRef]
- Rahantamalala, A.; Rech, P.; Martinez, Y.; Chaubet-Gigot, N.; Grima-Pettenati, J.; Pacquit, V. Coordinated Transcriptional Regulation of Two Key Genes in the Lignin Branch Pathway—CAD and CCR—Is Mediated through MYB- Binding Sites. BMC Plant Biol. 2010, 10, 130. [Google Scholar] [CrossRef]
- Zhong, R.; Morrison, W.H.; Himmelsbach, D.S.; Poole, F.L.; Ye, Z.-H. Essential Role of Caffeoyl Coenzyme A O-Methyltransferase in Lignin Biosynthesis in Woody Poplar Plants. Plant Physiol. 2000, 124, 563–578. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Tian, Y. STOP1 and STOP1-like Proteins, Key Transcription Factors to Cope with Acid Soil Syndrome. Front. Plant Sci. 2023, 14, 1200139. [Google Scholar] [CrossRef]
- Chen, C.; Galon, Y.; Rahmati Ishka, M.; Malihi, S.; Shimanovsky, V.; Twito, S.; Rath, A.; Vatamaniuk, O.K.; Miller, G. ASCORBATE PEROXIDASE6 Delays the Onset of Age-Dependent Leaf Senescence. Plant Physiol. 2021, 185, 441–456. [Google Scholar] [CrossRef] [PubMed]
- Breitel, D.A.; Chappell-Maor, L.; Meir, S.; Panizel, I.; Puig, C.P.; Hao, Y.; Yifhar, T.; Yasuor, H.; Zouine, M.; Bouzayen, M.; et al. AUXIN RESPONSE FACTOR 2 Intersects Hormonal Signals in the Regulation of Tomato Fruit Ripening. PLoS Genet. 2016, 12, e1005903. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Arai, K.; Aoi, Y.; Tanaka, Y.; Hira, H.; Guo, R.; Hu, Y.; Ge, C.; Zhao, Y.; Kasahara, H.; et al. The Main Oxidative Inactivation Pathway of the Plant Hormone Auxin. Nat. Commun. 2021, 12, 6752. [Google Scholar] [CrossRef]
- Hu, G.; Wang, B.; Jia, P.; Wu, P.; Lu, C.; Xu, Y.; Shi, L.; Zhang, F.; Zhong, N.; Chen, A.; et al. The Cotton miR530-SAP6 Module Activated by Systemic Acquired Resistance Mediates Plant Defense against Verticillium dahliae. Plant Sci. 2023, 330, 111647. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, L.-F.; Bhutto, S.H.; He, X.-R.; Yang, X.-M.; Zhou, X.-H.; Lin, X.-Y.; Rajput, A.A.; Li, G.-B.; Zhao, J.-H.; et al. Blocking miR530 Improves Rice Resistance, Yield, and Maturity. Front. Plant Sci. 2021, 12, 729560. [Google Scholar] [CrossRef]
- Cooley, M.B.; Pathirana, S.; Wu, H.J.; Kachroo, P.; Klessig, D.F. Members of the Arabidopsis HRT/RPP8 Family of Resistance Genes Confer Resistance to Both Viral and Oomycete Pathogens. Plant Cell 2000, 12, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.Y.; Lee, J.H.; Oh, C.-S.; Kang, H.-G.; Park, J.M. Endoplasmic Reticulum Stress Responses Function in the HRT-Mediated Hypersensitive Response in Nicotiana Benthamiana. Mol. Plant Pathol. 2016, 17, 1382–1397. [Google Scholar] [CrossRef]
- Dong, O.X.; Tong, M.; Bonardi, V.; El Kasmi, F.; Woloshen, V.; Wünsch, L.K.; Dangl, J.L.; Li, X. TNL-Mediated Immunity in Arabidopsis Requires Complex Regulation of the Redundant ADR1 Gene Family. New Phytol. 2016, 210, 960–973. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Tian, L.; Liu, X.; Zhang, Y.; Li, X. TIR Signal Promotes Interactions between Lipase-like Proteins and ADR1-L1 Receptor and ADR1-L1 Oligomerization. Plant Physiol. 2021, 187, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Y.; Liu, J.; Ding, Y.; Wang, S.; Zhang, X.; Liu, Y.; Yang, S. Temperature-dependent Autoimmunity Mediated by Chs1 Requires Its Neighboring TNL Gene SOC 3. New Phytol. 2017, 213, 1330–1345. [Google Scholar] [CrossRef] [PubMed]
- Pirrò, S.; Matic, I.; Guidi, A.; Zanella, L.; Gismondi, A.; Cicconi, R.; Bernardini, R.; Colizzi, V.; Canini, A.; Mattei, M.; et al. Identification of microRNAs and Relative Target Genes in Moringa Oleifera Leaf and Callus. Sci. Rep. 2019, 9, 15145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Mu, Y.; Zhang, H.; Yu, C. Maintenance of Stem Cell Activity in Plant Development and Stress Responses. Front. Plant Sci. 2023, 14, 1302046. [Google Scholar] [CrossRef]
- Beck, G.; Coman, D.; Herren, E.; Ruiz-Sola, M.A.; Rodríguez-Concepción, M.; Gruissem, W.; Vranová, E. Characterization of the GGPP Synthase Gene Family in Arabidopsis Thaliana. Plant Mol. Biol. 2013, 82, 393–416. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Read Data and Small RNA | Leaves | Stems | Roots |
|---|---|---|---|
| Raw reads | 37,535,006 | 36,751,657 | 25,226,124 |
| Clean, high-quality reads (16–40 bp) | 14,148,212 | 9,669,204 | 21,680,063 |
| rRNA | 1,574,564 | 2,269,843 | 6,389,370 |
| tRNA | 1,668,102 | 637,753 | 1,264,217 |
| Repeated Elements | 2,227,046 | 316,027 | 1,034,962 |
| snRNA | 9898 | 14,532 | 23,789 |
| snoRNA | 24,241 | 16,797 | 33,475 |
| scRNA | 25,396 | 28,810 | 446 |
| lncRNA | 50 | 14 | 103 |
| mRNA | 660,540 | 306,365 | 809,679 |
| miRNA | 335,241 | 720,488 | 16,1407 |
| Unaligned reads | 7,623,134 | 5,358,575 | 11,962,615 |
| Df | R2 | F | Pr (>F) | |
|---|---|---|---|---|
| Organs | 2 | 0.3375 | 1.528 | 0.013 |
| Residual | 6 | 0.6625 |
| miRNA | Target Genes and Functions |
|---|---|
| Novel_122 (Down) Leaf vs. Stem | -ARF2: Auxin Response Factor |
| -DWF4 (CYP90B1): Enzyme involved in the biosynthesis of brassinosteroids, a class of plant hormones derived from terpenoids. | |
| Novel_172 (Down) Leaf vs. Stem | -CCS: Copper Chaperone for Superoxide Dismutase (CCS) -RBGD2: RNA-binding glycine-rich protein -NHX4: Sodium/hydrogen exchanger protein -GH9A4: Member of the glycoside hydrolase family 9 (GH9) -AGL52: MADS-box transcription factor -A: PX6Ascorbate peroxidase isoform |
| Novel_189 (Down) Leaf vs. Stem | -LBD2: LATERAL ORGAN BOUNDARIES DOMAIN transcription factor -NDR1/HIN1-like: protein associated with plant defense responses, upregulated during pathogen attack. -SNM1: Involved in DNA repair of interstrand cross-links -CCoAOMT1: Involved in lignin biosynthesis -DXPS3/DXS: A key enzyme for isoprenoid biosynthesis -ILL3: Involved in auxin metabolism, regulating hormone homeostasis. |
| Novel_1 (Up) Leaf vs. Stem | -NPF5.5: Nitrate transporter, which is interconnected with isoprenoid biosynthesis pathways. -HB33/ZHD5: Involved in regulating gene expression crucial for maintaining the identity and function of the shoot apical meristem. |
| Novel_3 (Up) Leaf vs. Stem | -GET3a and GET3c: Components of the Guided Entry of Tail-anchored proteins, which is involved in targeting tail-anchored proteins to the endoplasmic reticulum membrane. -GGPPS3/GGPS4: Enzymes that produce geranylgeranyl diphosphate (GGPP), a precursor for various isoprenoids, including chlorophylls, carotenoids, and gibberellins. -SOC3: Involved in regulating flowering time. Also involved with maintaining the identity of the shoot apical meristem (GO:0010492). -HRT/RCY1/RPP8: Involved in plant immunity mediating defense responses against pathogens. -PLDDELTA: Phospholipase D delta enzyme. Role in stress responses and membrane remodeling. |
| Novel_31 (Up) Leaf vs. Stem | WAK4: Wall-Associated Kinase 4, member of the wall-associated kinase family. Involved in cell elongation and response to pathogens. RPG2/SWEET13: Sugars Will Eventually Be Exported Transporter 13, a member of the SWEET family of sugar transporters. Responds to biotic stress by modulating sugar transport (availability). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Estrada, R.; Rodriguez, L.; Romero, Y.; Arteaga, L.; Ruelas-Calloapaza, D.; Oha-Humpiri, F.; Flores, N.; Coila, P.; Arbizu, C.I. Profiling of Known and Novel microRNAs in an Oleaginous Crop Native to the Amazon Basin, Sacha Inchi (Plukenetia volubilis), Through smallRNA-Seq. Genes 2025, 16, 417. https://doi.org/10.3390/genes16040417
Estrada R, Rodriguez L, Romero Y, Arteaga L, Ruelas-Calloapaza D, Oha-Humpiri F, Flores N, Coila P, Arbizu CI. Profiling of Known and Novel microRNAs in an Oleaginous Crop Native to the Amazon Basin, Sacha Inchi (Plukenetia volubilis), Through smallRNA-Seq. Genes. 2025; 16(4):417. https://doi.org/10.3390/genes16040417
Chicago/Turabian StyleEstrada, Richard, Lila Rodriguez, Yolanda Romero, Linda Arteaga, Domingo Ruelas-Calloapaza, Filiberto Oha-Humpiri, Nils Flores, Pedro Coila, and Carlos I. Arbizu. 2025. "Profiling of Known and Novel microRNAs in an Oleaginous Crop Native to the Amazon Basin, Sacha Inchi (Plukenetia volubilis), Through smallRNA-Seq" Genes 16, no. 4: 417. https://doi.org/10.3390/genes16040417
APA StyleEstrada, R., Rodriguez, L., Romero, Y., Arteaga, L., Ruelas-Calloapaza, D., Oha-Humpiri, F., Flores, N., Coila, P., & Arbizu, C. I. (2025). Profiling of Known and Novel microRNAs in an Oleaginous Crop Native to the Amazon Basin, Sacha Inchi (Plukenetia volubilis), Through smallRNA-Seq. Genes, 16(4), 417. https://doi.org/10.3390/genes16040417