
Genetic Testing in Adults over 50 Years with Chronic Kidney Disease: Diagnostic Yield and Clinical Implications in a Specialized Kidney Genetics Clinic

 , , , , , , , , and add
Show full author list
, , , , , , , , and add
Show full author list
Abstract
1. Introduction
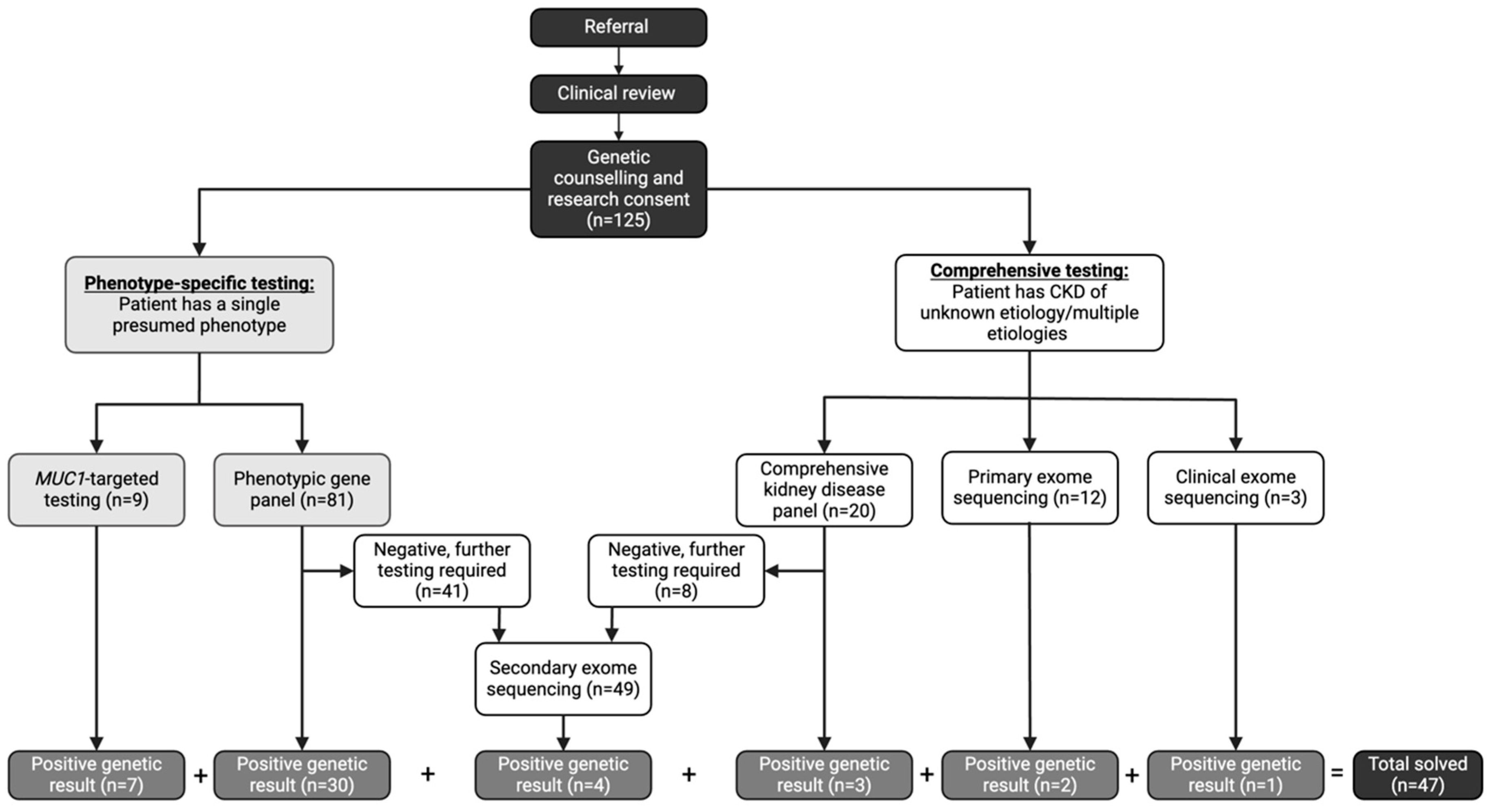
2. Methods
- (1)
- Phenotypic-driven gene panel testing which includes genes specific for a certain subtype of disease (i.e., cystic kidney disease). This is the first-line approach when a specific CKD subtype is suspected.
- (2)
- Comprehensive inherited kidney disease gene panel which includes 331 genes known to cause CKD across different phenotypes. This approach is employed when the phenotype is ambiguous or multiple overlapping conditions are suspected.
- (3)
- MUC1-targeted gene testing for the cytosine insertion in the mucin-1 (MUC1) gene in participants with high clinical suspicion of autosomal dominant tubulo-interstitial kidney disease (ADTKD) (i.e., non-proteinuric CKD with onset ≥18 years, family pedigree consistent with an autosomal dominant mode of inheritance, bland urine sediment at initial diagnosis of CKD, and personal or family history of gout) [19]. This testing is not currently available on any commercial sequencing platforms; therefore, the testing was performed in the Clinical Laboratory Improvement Amendments (CLIA) certified laboratory at the Broad Institute and Massachusetts Institute of Technology. This targeted testing is required due to the high guanosine-cytosine content and repetitive nature of the 60-mer variable number of tandem repeat (VNTR) sequences [20].
- (4)
- Clinical exome sequencing is employed as a first line test in eligible participants who met the Genome-Wide Sequencing Ontario criteria for testing which included the following: moderate to severe developmental or functional impairment, multisystem involvement, progressive clinical course, differential diagnosis required multiple targeted gene panels, and suspected severe genetic syndrome for which multiple family member are affected or where parents are consanguineous [21].
- (5)
- Research exome sequencing assessing for all implicated causes of genetic CKD (694 genes) using a virtual, bioinformatic panel approach [22] in individuals who did not meet current clinical criteria for clinical exome sequencing (primary) or who remained undiagnosed after panel testing (secondary). This exome sequencing is performed in a research laboratory with findings confirmed in a CLIA certified laboratory through specific mutation confirmation, as previously described [14].
2.1. Primary Outcome
2.2. Statistical Analysis
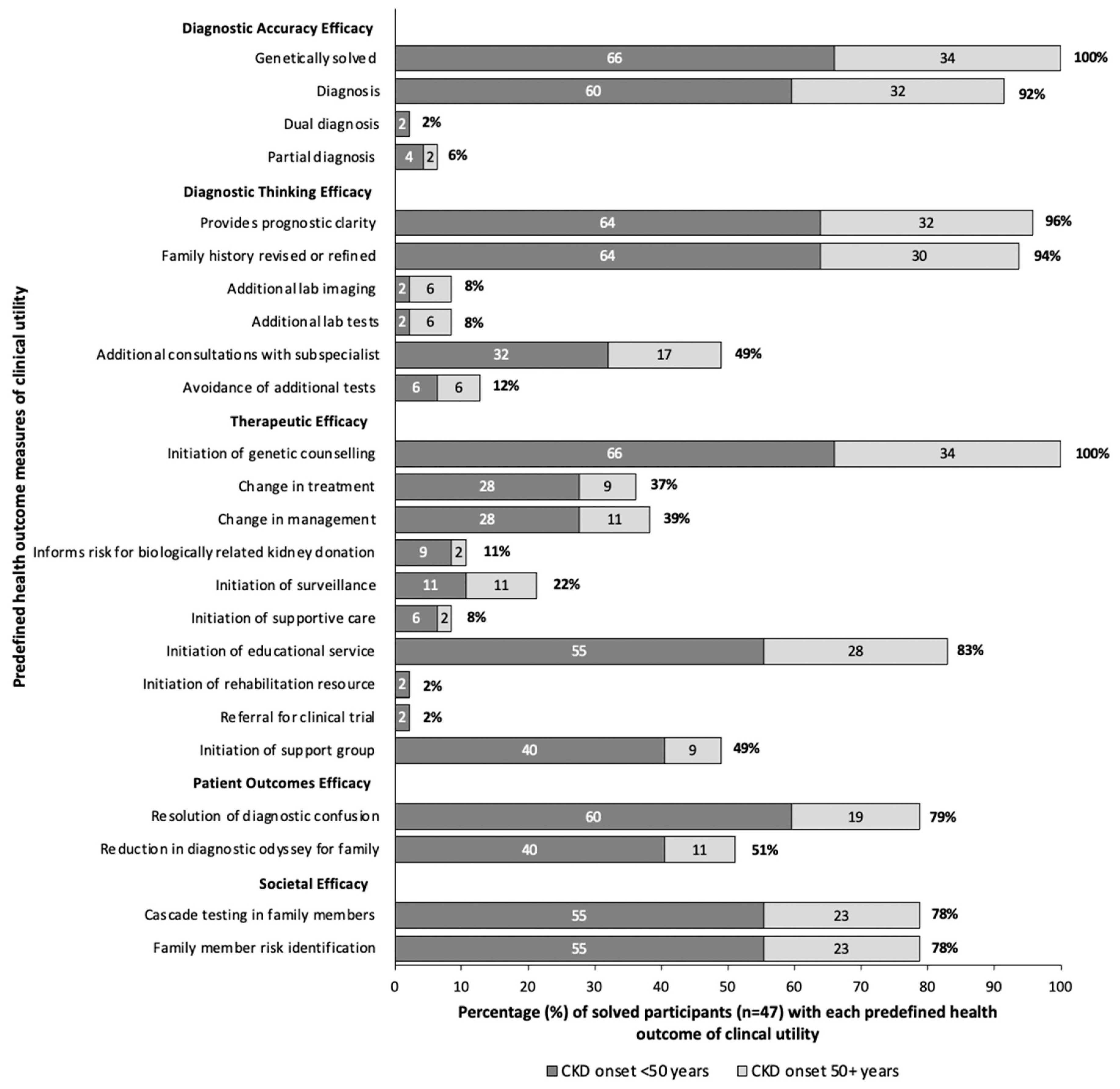
2.3. Health Outcomes Related to Clinical Utility
2.4. Case Descriptions
3. Results
3.1. Demographics
3.2. Diagnostic Yield
3.3. Risk Factors for Genetic Disease
3.4. Time to Diagnosis
3.5. Clinical Utility
4. Case Descriptions
4.1. Case 1. X-Linked Hypophosphatemia
4.1.1. Patient History
4.1.2. Genetic Testing, Diagnosis, and Pathophysiology
4.1.3. Clinical Implications and Management
4.2. Case 2. Mitochondrial Cytopathies
4.2.1. Patient History
4.2.2. Genetic Testing and Diagnosis
4.2.3. Clinical Implications and Management
4.3. Cases 3 and 4. Atypical Cystic Kidney Disease
4.3.1. Patient Histories
4.3.2. Genetic Insights into Cystic Kidney Disease
4.3.3. Clinical Implications and Importance of Genetic Diagnosis
4.4. Case 5. Congenital Thrombotic Thrombocytopenia Purpura
4.4.1. Patient History
4.4.2. Clinical Implications
4.5. Autosomal Dominant Tubulo-Interstitial Kidney Disease
Diagnostic Odyssey in ADTKD
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dahl, N.K.; Bloom, M.S.; Chebib, F.T.; Clark, D.; Westemeyer, M.; Jandeska, S.; Zhang, Z.; Milo-Rasouly, H.; Kolupaeva, V.; Marasa, M.; et al. The Clinical Utility of Genetic Testing in the Diagnosis and Management of Adults with Chronic Kidney Disease. J. Am. Soc. Nephrol. 2023, 34, 2039. [Google Scholar] [CrossRef] [PubMed]
- Groopman, E.E.; Marasa, M.; Cameron-Christie, S.; Petrovski, S.; Aggarwal, V.S.; Milo-Rasouly, H.; Li, Y.; Zhang, J.; Nestor, J.; Krithivasan, P.; et al. Diagnostic Utility of Exome Sequencing for Kidney Disease. N. Engl. J. Med. 2019, 380, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Abdulrahim, J.W.; Kwee, L.C.; Alenezi, F.; Sun, A.Y.; Baras, A.; Ajayi, T.A.; Henao, R.; Holley, C.; McGarrah, R.; Daubert, J.P.; et al. Identification of Undetected Monogenic Cardiovascular Disorders. J. Am. Coll. Cardiol. 2020, 76, 797–808. [Google Scholar] [CrossRef]
- Van Der Lee, S.J.; Hulsman, M.; Van Spaendonk, R.; Van Der Schaar, J.; Dijkstra, J.; Tesi, N.; van Ruissen, F.; Elting, M.; Reinders, M.; De Rojas, I.; et al. Prevalence of Pathogenic Variants and Eligibility Criteria for Genetic Testing in Patients Who Visit a Memory Clinic. Neurology 2025, 104, e210273. [Google Scholar] [CrossRef]
- Ganapathy, A.; Mishra, A.; Soni, M.R.; Kumar, P.; Sadagopan, M.; Kanthi, A.V.; Patric, I.R.P.; George, S.; Sridharan, A.; Thyagarajan, T.C.; et al. Multi-gene testing in neurological disorders showed an improved diagnostic yield: Data from over 1000 Indian patients. J. Neurol. 2019, 266, 1919–1926. [Google Scholar] [CrossRef]
- Schott, C.; Lebedeva, V.; Taylor, C.; Abumelha, S.; Roshanov, P.S.; Connaughton, D.M. Utility of Genetic Testing in Adults with Chronic Kidney Disease: A Systematic Review and Meta-Analysis. Clin. J. Am. Soc. Nephrol. 2024, 20, 101–115. [Google Scholar] [CrossRef]
- Elliott, M.D.; James, L.C.; Simms, E.L.; Sharma, P.; Girard, L.P.; Cheema, K.; Elliott, M.J.; Lauzon, J.L.; Chun, J. Mainstreaming Genetic Testing for Adult Patients With Autosomal Dominant Polycystic Kidney Disease. Can. J. Kidney Health Dis. 2021, 8, 20543581211055001. [Google Scholar] [CrossRef] [PubMed]
- Ames, E.G.; Anand, P.M.; Bekheirnia, M.R.; Doshi, M.D.; El Ters, M.; Freese, M.E.; Gbadegesin, R.A.; Guay-Woodford, L.M.; Java, A.; Ranch, D.; et al. Evaluation for genetic disease in kidney transplant candidates: A practice resource. Am. J. Transplant. 2025, 25, 237–249. [Google Scholar] [CrossRef]
- Aron, A.W.; Dahl, N.K.; Besse, W. A Practical Guide to Genetic Testing for Kidney Disorders of Unknown Etiology. Kidney360 2022, 3, 1640–1651. [Google Scholar] [CrossRef]
- National Health Services England. National Genomic Test Directory Testing Criteria for Rare and Inherited Disease. 2024. Available online: https://www.england.nhs.uk/wp-content/uploads/2024/07/national-genomic-test-directory-rare-and-inherited-disease-eligibility-criteria-v7.pdf (accessed on 26 March 2024).
- Guidelines for Genetic Diagnostics in Kidney Disease; MEDonline Publisher: Breukelen, The Netherlands, 2018; Available online: https://publicatie.nefro.nl/richtlijnen/handreiking-genetische-diagnostiek-bij-nierziekten/ (accessed on 17 December 2024).
- Snoek, R.; van Jaarsveld, R.H.; Nguyen, T.Q.; Peters, E.D.J.; Elferink, M.G.; Ernst, R.F.; Rookmaaker, M.B.; Lilien, M.R.; Spierings, E.; Goldschmeding, R.; et al. Genetics-first approach improves diagnostics of ESKD patients <50 years old. Nephrol. Dial. Transplant. 2022, 37, 349–357. [Google Scholar] [CrossRef]
- Vivante, A.; Hildebrandt, F. Exploring the genetic basis of early-onset chronic kidney disease. Nat. Rev. Nephrol. 2016, 12, 133–146. [Google Scholar] [CrossRef]
- Schott, C.; Arnaldi, M.; Baker, C.; Wang, J.; McIntyre, A.D.; Colaicovo, S.; Relouw, S.; Offerni, G.A.; Campagnolo, C.; Nyatten, L.R.V.; et al. Implementation of a kidney genetic service into the diagnostic pathway for patients with chronic kidney disease in Canada. Kidney Int. Rep. 2025, 10, 574–590. [Google Scholar] [CrossRef] [PubMed]
- KDIGO Conference Participants. Genetics in chronic kidney disease: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2022, 101, 1126–1141. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, F. Genetic kidney diseases. Lancet 2010, 375, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Merchant, A.A.; Ling, E. An approach to treating older adults with chronic kidney disease. CMAJ 2023, 195, E612–E618. [Google Scholar] [CrossRef]
- Levey, A.S.; Eckardt, K.-U.; Tsukamoto, Y.; Levin, A.; Coresh, J.; Rossert, J.; Zeeuw, D.D.E.; Hostetter, T.H.; Lameire, N.; Eknoyan, G. Definition and classification of chronic kidney disease: A position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int. 2005, 67, 2089–2100. [Google Scholar] [CrossRef]
- Ayasreh, N.; Bullich, G.; Miquel, R.; Furlano, M.; Ruiz, P.; Lorente, L.; Valero, O.; García-González, M.A.; Arhda, N.; Garin, I.; et al. Autosomal Dominant Tubulointerstitial Kidney Disease: Clinical Presentation of Patients With ADTKD-UMOD and ADTKD-MUC1. Am. J. Kidney Dis. 2018, 72, 411–418. [Google Scholar] [CrossRef]
- Blumenstiel, B.; DeFelice, M.; Birsoy, O.; Bleyer, A.J.; Kmoch, S.; Carter, T.A.; Gnirke, A.; Kidd, K.; Rehm, H.L.; Ronco, L.; et al. Development and Validation of a Mass Spectrometry-Based Assay for the Molecular Diagnosis of Mucin-1 Kidney Disease. J. Mol. Diagn. 2016, 18, 566–571. [Google Scholar] [CrossRef]
- Szuto, A. Genome-Wide Sequencing Ontario. 2023. Provincial Update: Exome Sequencing for Rare Disease Diagnostics. Available online: https://gsontario.ca/for-providers/patient-eligibility/ (accessed on 15 July 2024).
- Sheikh Hassani, M.; Jain, R.; Ramaswamy, S.; Sinha, S.; El Naofal, M.; Halabi, N.; Alyafei, S.; Alfalasi, R.; Shenbagam, S.; Taylor, A.; et al. Virtual Gene Panels Have a Superior Diagnostic Yield for Inherited Rare Diseases Relative to Static Panels. Clin. Chem. 2025, 71, 169–184. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Hayeems, R.Z.; Dimmock, D.; Bick, D.; Belmont, J.W.; Green, R.C.; Lanpher, B.; Jobanputra, V.; Mendoza, R.; Kulkarni, S.; Grove, M.E.; et al. Clinical utility of genomic sequencing: A measurement toolkit. NPJ Genom. Med. 2020, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Ariceta, G.; Beck-Nielsen, S.S.; Boot, A.M.; Brandi, M.L.; Briot, K.; de Lucas Collantes, C.; Emma, F.; Giannini, S.; Haffner, D.; Keen, R.; et al. The International X-Linked Hypophosphatemia (XLH) Registry: First interim analysis of baseline demographic, genetic and clinical data. Orphanet J. Rare Dis. 2023, 18, 304. [Google Scholar] [CrossRef]
- Pettifor, J.M. What’s new in hypophosphataemic rickets? Eur. J. Pediatr. 2008, 167, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Kayser, M.; Jain, P.; Bale, A.; Carpenter, T.O. A De Novo Deleterious PHEX Variant Without Clinical Features of X-Linked Hypophosphatemia. JCEM Case Rep. 2023, 1, luad082. [Google Scholar] [CrossRef] [PubMed]
- Giannini, S.; Bianchi, M.L.; Rendina, D.; Massoletti, P.; Lazzerini, D.; Brandi, M.L. Burden of disease and clinical targets in adult patients with X-linked hypophosphatemia. A comprehensive review. Osteoporos. Int. 2021, 32, 1937–1949. [Google Scholar] [CrossRef]
- Haffner, D.; Emma, F.; Eastwood, D.M.; Duplan, M.B.; Bacchetta, J.; Schnabel, D.; Wicart, P.; Bockenhauer, D.; Santos, F.; Levtchenko, E.; et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat. Rev. Nephrol. 2019, 15, 435. [Google Scholar] [CrossRef]
- Emma, F.; Salviati, L. Mitochondrial cytopathies and the kidney. Nephrol. Ther. 2017, 13 (Suppl. S1), S23–S28. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.S.L.; Longley, M.J.; Copeland, W.C. The common A467T mutation in the human mitochondrial DNA polymerase (POLG) compromises catalytic efficiency and interaction with the accessory subunit. J. Biol. Chem. 2005, 280, 31341–31346. [Google Scholar] [CrossRef]
- Parikh, S.; Goldstein, A.; Karaa, A.; Koenig, M.K.; Anselm, I.; Brunel-Guitton, C.; Christodoulou, J.; Cohen, B.H.; Dimmock, D.; Enns, G.M.; et al. Patient care standards for primary mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2017, 19, 1380. [Google Scholar] [CrossRef]
- Cohen, B.H.; Chinnery, P.F.; Copeland, W.C. POLG-Related Disorders. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK26471/ (accessed on 28 May 2024).
- Cornec-Le Gall, E.; Alam, A.; Perrone, R.D. Autosomal dominant polycystic kidney disease. Lancet 2019, 393, 919–935. [Google Scholar] [CrossRef]
- Fujimaru, T.; Mori, T.; Sekine, A.; Mandai, S.; Chiga, M.; Kikuchi, H.; Ando, F.; Mori, Y.; Nomura, N.; Iimori, S.; et al. Kidney enlargement and multiple liver cyst formation implicate mutations in PKD1/2 in adult sporadic polycystic kidney disease. Clin. Genet. 2018, 94, 125–131. [Google Scholar] [CrossRef]
- Senum, S.R.; Li, Y.M.; Benson, K.A.; Joli, G.; Olinger, E.; Lavu, S.; Madsen, C.D.; Gregory, A.V.; Neatu, R.; Kline, T.L.; et al. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney-spectrum phenotype. Am. J. Hum. Genet. 2022, 109, 136–156. [Google Scholar] [CrossRef]
- Nuñez Zuno, J.A.; Khaddour, K. Thrombotic Thrombocytopenic Purpura Evaluation and Management. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK470585/ (accessed on 10 July 2024).
- Tsai, H.-M. The kidney in thrombotic thrombocytopenic purpura. Minerva Med. 2007, 98, 731–747. [Google Scholar] [PubMed]
- Dorgalaleh, A.; Mahmudi, M.; Tabibian, S.; Khatib, Z.K.; Tamaddon, G.H.; Moghaddam, E.S.; Bamedi, T.; Alizadeh, S.; Moradi, E. Anemia and Thrombocytopenia in Acute and Chronic Renal Failure. Int. J. Hematol.-Oncol. Stem Cell Res. 2013, 7, 34–39. [Google Scholar] [PubMed]
- Živná, M.; Kidd, K.O.; Barešová, V.; Hůlková, H.; Kmoch, S.; Bleyer, A.J. Autosomal dominant tubulointerstitial kidney disease: A review. Am. J. Med. Genet. C Semin. Med. Genet. 2022, 190, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Kmochová, T.; Kidd, K.O.; Orr, A.; Hnízda, A.; Hartmannová, H.; Hodaňová, K.; Vyleťal, P.; Naušová, K.; Brinsa, V.; Trešlová, H.; et al. Autosomal dominant ApoA4 mutations present as tubulointerstitial kidney disease with medullary amyloidosis. Kidney Int. 2024, 105, 799–811. [Google Scholar] [CrossRef]
- Rasouly, H.M.; Balderes, O.; Marasa, M.; Fernandez, H.; Lipton, M.; Lin, F.; Gharavi, A.G.; Sabatello, M. The effect of genetic education on the referral of patients to genetic evaluation: Findings from a national survey of nephrologists. Genet. Med. 2023, 25, 100814. [Google Scholar] [CrossRef]
- Mrug, M.; Bloom, M.S.; Seto, C.; Malhotra, M.; Tabriziani, H.; Gauthier, P.; Sidlow, V.; McKanna, T.; Billings, P.R. Genetic Testing for Chronic Kidney Diseases: Clinical Utility and Barriers Perceived by Nephrologists. Kidney Med. 2021, 3, 1050–1056. [Google Scholar] [CrossRef]
- Dvela-Levitt, M.; Kost-Alimova, M.; Emani, M.; Kohnert, E.; Thompson, R.; Sidhom, E.-H.; Rivadeneira, A.; Sahakian, N.; Roignot, J.; Papagregoriou, G.; et al. Small Molecule Targets TMED9 and Promotes Lysosomal Degradation to Reverse Proteinopathy. Cell 2019, 178, 521–535.e23. [Google Scholar] [CrossRef]
- Jayasinghe, K.; Quinlan, C.; Mallett, A.J.; Kerr, P.G.; McClaren, B.; Nisselle, A.; Mallawaarachchi, A.; Polkinghorne, K.R.; Patel, C.; Best, S.; et al. Attitudes and Practices of Australian Nephrologists Toward Implementation of Clinical Genomics. Kidney Int. Rep. 2021, 6, 272–283. [Google Scholar] [CrossRef]
- Connaughton, D.M.; Kennedy, C.; Shril, S.; Mann, N.; Murray, S.L.; Williams, P.A.; Conlon, E.; Nakayama, M.; van der Ven, A.T.; Ityel, H.; et al. Monogenic causes of chronic kidney disease in adults. Kidney Int. 2019, 95, 914–928. [Google Scholar] [CrossRef] [PubMed]
- Lata, S.; Marasa, M.; Li, Y.; Fasel, D.A.; Groopman, E.; Jobanputra, V.; Rasouly, H.; Mitrotti, A.; Westland, R.; Verbitsky, M.; et al. Whole-Exome Sequencing in Adults With Chronic Kidney Disease A Pilot Study. Ann. Intern. Med. 2018, 168, 100–109. [Google Scholar] [CrossRef]
- Braun, D.A.; Schueler, M.; Halbritter, J.; Gee, H.Y.; Porath, J.D.; Lawson, J.A.; Airik, R.; Shril, S.; Allen, S.J.; Stein, D.; et al. Whole exome sequencing identifies causative mutations in the majority of consanguineous or familial cases with childhood-onset increased renal echogenicity. Kidney Int. 2016, 89, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Burnes, D.; Sheppard, C.; Henderson, C.R.; Wassel, M.; Cope, R.; Barber, C.; Pillemer, K. Interventions to Reduce Ageism Against Older Adults: A Systematic Review and Meta-Analysis. Am. J. Public Health 2019, 109, e1–e9. [Google Scholar] [CrossRef]
- Beal, F.; Forrester, N.; Watson, E.; Williams, M.; Buckton, A.; Marlais, M.; Maxted, A.; UK Gene Panel Study Group; Woolf, A.S.; Saleem, M.A.; et al. A targeted gene panel illuminates pathogenesis in young people with unexplained kidney failure. J. Nephrol. 2024, 37, 1273–1284. [Google Scholar] [CrossRef]
- Knoers, N.; Antignac, C.; Bergmann, C.; Dahan, K.; Giglio, S.; Heidet, L.; Lipska-Ziętkiewicz, B.S.; Noris, M.; Remuzzi, G.; Vargas-Poussou, R.; et al. Genetic testing in the diagnosis of chronic kidney disease: Recommendations for clinical practice. Nephrol. Dial. Transplant. 2022, 37, 239–254. [Google Scholar] [CrossRef]
- Bleyer, A.J.; Kmoch, S.; Antignac, C.; Robins, V.; Kidd, K.; Kelsoe, J.R.; Hladik, G.; Klemmer, P.; Knohl, S.J.; Scheinman, S.J.; et al. Variable clinical presentation of an MUC1 mutation causing medullary cystic kidney disease type 1. Clin. J. Am. Soc. Nephrol. 2014, 9, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Dusic, E.J.; Theoryn, T.; Wang, C.; Swisher, E.M.; Bowen, D.J. Barriers, interventions, and recommendations: Improving the genetic testing landscape. Front. Digit. Health 2022, 4, 961128. [Google Scholar] [CrossRef]
- Schott, C.; Dilliott, A.A.; Wang, J.; McIntyre, A.D.; Son, S.; Colaiacovo, S.; Baker, C.; Gunaratnam, L.; House, A.A.; Huang, S.-H.S.; et al. Vascular calcification in chronic kidney disease associated with pathogenic variants in ABCC6. Gene 2024, 927, 148731. [Google Scholar]
- Schott, C.; Colaiacovo, S.; Baker, C.; Weir, M.A.; Connaughton, D.M. Reclassification of Genetic Testing Results: A Case Report Demonstrating the Need for Structured Re-Evaluation of Genetic Findings. Can. J. Kidney Health Dis. 2024, 11, 20543581241242562. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Total Participants (n (Proportion in %)) | Solved Participants (n) | Unsolved Participants (n) | Diagnostic Yield (%) | p Value | |
|---|---|---|---|---|---|
| Total participants | 125 | 47 | 78 | 38 | |
| Total families | 114 | 40 | 74 | 35 | |
| Sex | |||||
| Male | 59 (47) | 20 | 39 | 33 | 0.53 |
| Female | 66 (53) | 27 | 39 | 41 | |
| Age at testing | |||||
| 50–54 | 21 (17) | 10 | 11 | 48 | 0.43 |
| 55–59 | 25 (20) | 11 | 14 | 44 | 0.61 |
| 60–64 | 23 (18) | 9 | 14 | 39 | 1.00 |
| 65–69 | 26 (21) | 9 | 17 | 35 | 0.11 |
| 70–74 | 13 (10) | 3 | 10 | 23 | 0.37 † |
| 75+ | 17 (14) | 5 | 12 | 29 | 0.63 |
| Race | |||||
| Asian | 8 (7) | 0 | 8 | 0 | 0.02 †* |
| Black | 6 (5) | 4 | 2 | 67 | 0.20 † |
| Hispanic | 3 (2) | 3 | 0 | 100 | 0.05 †* |
| Indigenous | 4 (3) | 0 | 4 | 0 | 0.17 † |
| White | 80 (64) | 34 | 46 | 43 | 0.19 |
| Unknown | 24 (19) | 6 | 18 | 25 | 0.24 |
| Age at CKD diagnosis | |||||
| <18 | 7 (6) | 3 | 4 | 42 | 1.00 † |
| 19–29 | 15 (12) | 4 | 11 | 22 | 0.41 † |
| 30–39 | 18 (14) | 14 | 4 | 78 | 0.0002 †* |
| 40–49 | 22 (18) | 10 | 12 | 46 | 0.55 |
| 50–59 | 28 (24) | 8 | 20 | 29 | 0.37 |
| 60+ | 32 (26) | 8 | 24 | 25 | 0.14 |
| Unknown | 3 (2) | 0 | 3 | 0 | 0.29 † |
| CKD onset (known) | |||||
| 50+ years | 60 (49) | 16 | 44 | 27 | 0.06 ◊ |
| <50 years | 62 (51) | 31 | 31 | 50 | |
| ESKD | |||||
| Present | 48 (38) | 23 | 25 | 48 | 0.14 ◊◊ |
| Absent | 77 (62) | 24 | 53 | 31 | |
| Age at ESKD onset | |||||
| 50+ years | 25 (52) | 10 | 15 | 40 | 0.39 |
| <50 years | 23 (48) | 13 | 10 | 57 | |
| CKD etiology | |||||
| Unknown | 61 (49) | 22 | 39 | 36 | 0.86 |
| Presumed | 64 (51) | 25 | 39 | 39 | |
| Etiology if presumed | |||||
| ADTKD | 1 (1) | 1 | 0 | 100 | 0.39 † |
| CAKUT | 5 (8) | 1 | 4 | 20 | 0.64 † |
| Cystic kidney disease | 22 (34) | 11 | 11 | 50 | 0.30 |
| Diabetic nephropathy | 7 (11) | 2 | 5 | 29 | 0.70 † |
| Glomerulopathies | 12 (19) | 4 | 8 | 33 | 0.75 † |
| Hypertensive nephropathy | 10 (16) | 4 | 6 | 40 | 1.00 † |
| Stones disease | 5 (8) | 2 | 3 | 40 | 1.00 † |
| Tubulopathies | 2 (3) | 0 | 2 | 0 | 0.52 † |
| Family history | |||||
| Positive | 92 (74) | 40 | 52 | 43 | 0.03 *◊◊◊ |
| Negative | 33 (26) | 7 | 26 | 21 | |
| Extrarenal Features | |||||
| Positive | 78 (62) | 31 | 47 | 40 | 0.65 |
| Negative | 47 (38) | 16 | 31 | 34 | |
| Type of extrarenal feature | |||||
| Hearing loss | 25 (32) | 13 | 12 | 52 | 0.20 |
| Onset < 50 years | 11 (48) | 8 | 3 | 73 | 0.02 †* |
| Onset ≥ 50 years | 13 (52) | 5 | 8 | 38 | 0.84 |
| Gout | 35 (45) | 11 | 24 | 31 | 0.26 |
| Proceeding CKD | 12 (34) | 4 | 8 | 33 | 0.75 † |
| Following CKD | 23 (66) | 7 | 16 | 30 | 0.41 |
| Valvular heart disease | 1 (1) | 0 | 1 | 0 | 1.00 † |
| Hypospadias | 1 (1) | 0 | 1 | 0 | 1.00 † |
| Neurological disorder | 6 (8) | 2 | 4 | 33 | 1.00 † |
| Liver disease | 13 (17) | 8 | 5 | 62 | 0.15 |
| Cystic liver | 7 (54) | 6 | 1 | 86 | 0.01 †* |
| Fatty liver | 5 (39) | 2 | 3 | 40 | 1.00 † |
| Other | 1 (8) | 0 | 1 | 0 | 1.00 † |
| Eye pathology | 28 (36) | 8 | 20 | 29 | 0.20 |
| Testing type | |||||
| Comprehensive gene panel | 20 (12) | 3 | 17 | 15 | 0.29 † |
| Phenotypic gene panel | 81 (47) | 30 | 51 | 37 | 0.009 * |
| Clinical exome | 3 (2) | 1 | 2 | 33 | 1.00 † |
| Primary exome | 12 (7) | 2 | 10 | 17 | 0.52 † |
| Secondary exome | 49 (28) | 4 | 45 | 8 | 0.0005 †* |
| MUC1-specific testing | 9 (5) | 7 | 2 | 78 | 0.002 †* |
| Phenotype | Genotype | Percent of Solved Patients |
|---|---|---|
| Glomerulopathies including collagenopathies (14) | COL4A3 (6), APOL1 (3), COL4A5 (3), COL4A4 (2) | 30% |
| Tubulointerstitial kidney disease (11) | MUC1 (7), UMOD (4) | 24% |
| Cystic kidney disease (9) | PKD1 (5), IFT140 (2), ALG5 (1), PKD2 (1) | 20% |
| Mitochondrial and Rhabdomyolysis (4) | POLG (2), CPT2 (1), MTTL1 (1) | 9% |
| Vascular kidney disease including hypertension (3) | ABCC6 (3) | 7% |
| Nephrocalcinosis and nephrolithiasis (2) | SLC2A9 (1), SLC3A1 (1) | 4% |
| Complement/aHUS and other (2) | ADAMTS13 (1), CFH (1) | 4% |
| Tubulopathies (1) | PHEX (1) | 2% |
| Amyloidosis (1) | FGA (1) | 2% |
| Feature | Diagnostic Yield in Our Study | Recommendation |
|---|---|---|
| Early-onset CKD (<50 years) | 50% | Strongly consider testing |
| Family history of CKD | 43% | Strongly consider testing |
| Extrarenal features | 40% | Consider testing |
| CKD of unknown etiology | 36% | Consider testing |
| Clinical Outcome | Specific Outcome |
|---|---|
| Avoidance of certain medications |
|
| Referral to subspecialists for extrarenal a feature of disease |
|
| Use of additional lab images |
|
| Surveillance |
|
| Family # | Patient # | Presumed Etiology Pre-Testing | Age CKD/ESKD Onset Years | Age at MUC1 Diagnosis Years | Family History | Extrarenal Features | Hematuria, Proteinuria | Genetic Testing Strategy |
|---|---|---|---|---|---|---|---|---|
| F0006 | P0007 | Atypical cystic kidney disease | 48/54 | 57 | + | Macular degeneration | Hematuria | Cystic kidney disease panel, 2° exome sequencing, MUC1-targeted testing |
| P0306 | ADTKD | 34/60 | 63 | + | Gout | Bland urine | MUC1-targeted testing | |
| F0022 | P0027 | CKDu | 31/43 | 55 | + | Hyperpara-thyroidism | Bland urine | FSGS and HHL panels *, MUC1-targeted testing |
| P0050 | Hereditary nephritis | 38/43 | 65 | + | Hearing loss, hypertension | Bland urine | FSGS and HHL panels, MUC1-targeted testing | |
| F0303 | P0442 | CKDu | 42/53 | 62 | + | None | Hematuria, proteinuria | MUC1-targeted testing |
| P0504 | CKDu | 30/42 | 59 | + | None | Proteinuria | MUC1-targeted testing | |
| P0516 | CKDu | 37/37 | 54 | + | None | Bland urine | MUC1-targeted testing |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schott, C.; Alajmi, M.; Bukhari, M.; Relouw, S.; Wang, J.; McIntyre, A.D.; Baker, C.; Colaiacovo, S.; Campagnolo, C.; Almada Offerni, G.; et al. Genetic Testing in Adults over 50 Years with Chronic Kidney Disease: Diagnostic Yield and Clinical Implications in a Specialized Kidney Genetics Clinic. Genes 2025, 16, 408. https://doi.org/10.3390/genes16040408
Schott C, Alajmi M, Bukhari M, Relouw S, Wang J, McIntyre AD, Baker C, Colaiacovo S, Campagnolo C, Almada Offerni G, et al. Genetic Testing in Adults over 50 Years with Chronic Kidney Disease: Diagnostic Yield and Clinical Implications in a Specialized Kidney Genetics Clinic. Genes. 2025; 16(4):408. https://doi.org/10.3390/genes16040408
Chicago/Turabian StyleSchott, Clara, Mohammad Alajmi, Mohammad Bukhari, Sydney Relouw, Jian Wang, Adam D. McIntyre, Cadence Baker, Samantha Colaiacovo, Carla Campagnolo, Gabriela Almada Offerni, and et al. 2025. "Genetic Testing in Adults over 50 Years with Chronic Kidney Disease: Diagnostic Yield and Clinical Implications in a Specialized Kidney Genetics Clinic" Genes 16, no. 4: 408. https://doi.org/10.3390/genes16040408
APA StyleSchott, C., Alajmi, M., Bukhari, M., Relouw, S., Wang, J., McIntyre, A. D., Baker, C., Colaiacovo, S., Campagnolo, C., Almada Offerni, G., Blake, P. G., Chiu, M., Cowan, A., Garg, A. X., Gunaratnam, L., House, A. A., Huang, S.-H. S., Iyer, H., Jain, A. K., ... Connaughton, D. M. (2025). Genetic Testing in Adults over 50 Years with Chronic Kidney Disease: Diagnostic Yield and Clinical Implications in a Specialized Kidney Genetics Clinic. Genes, 16(4), 408. https://doi.org/10.3390/genes16040408