
SYNGAP1 Syndrome and the Brain Gene Registry

{kind=link}
{kind=link}
Abstract
1. Introduction
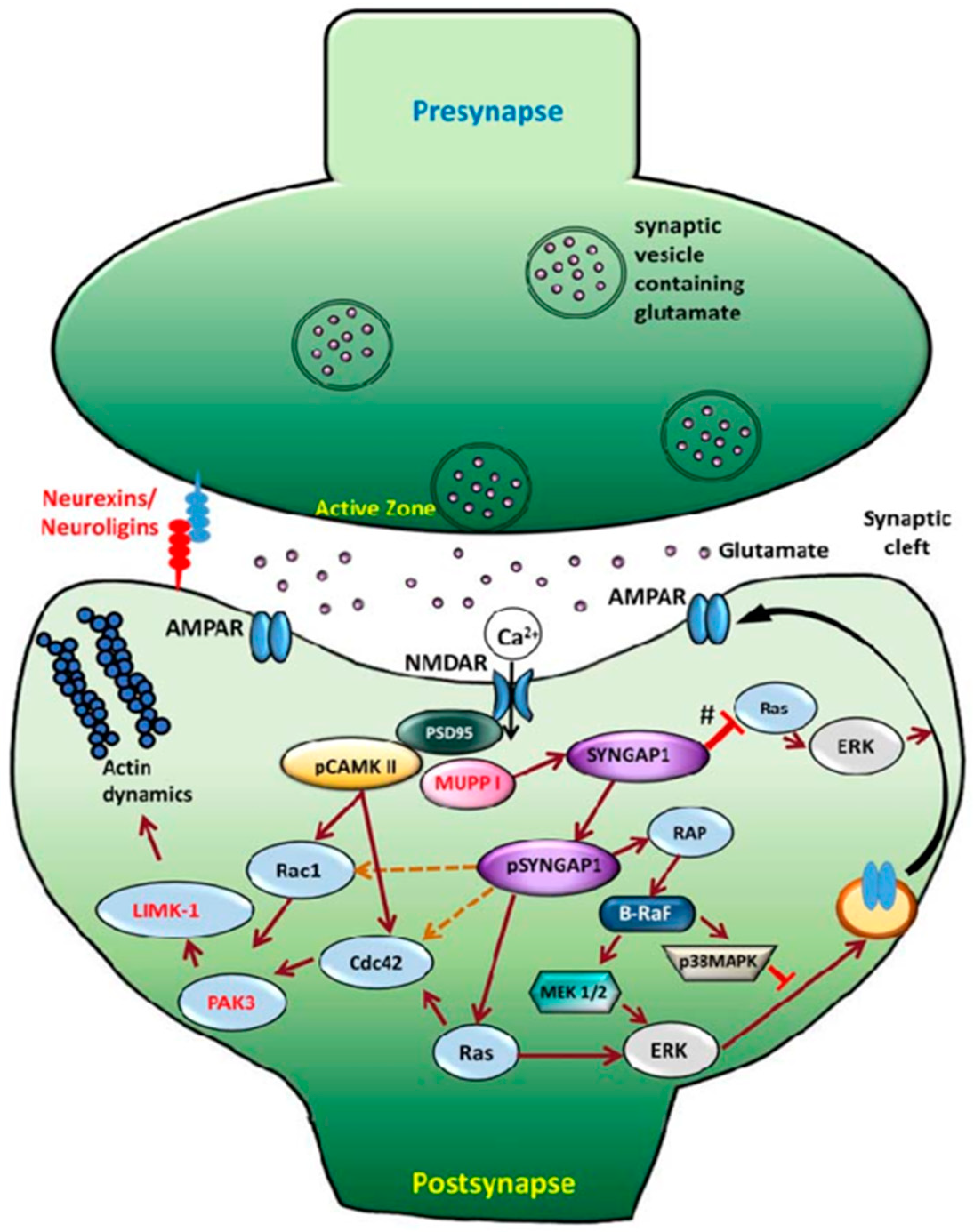
1.1. Review of SYNGAP1
1.2. Genotype and Phenotype
1.3. Treatment
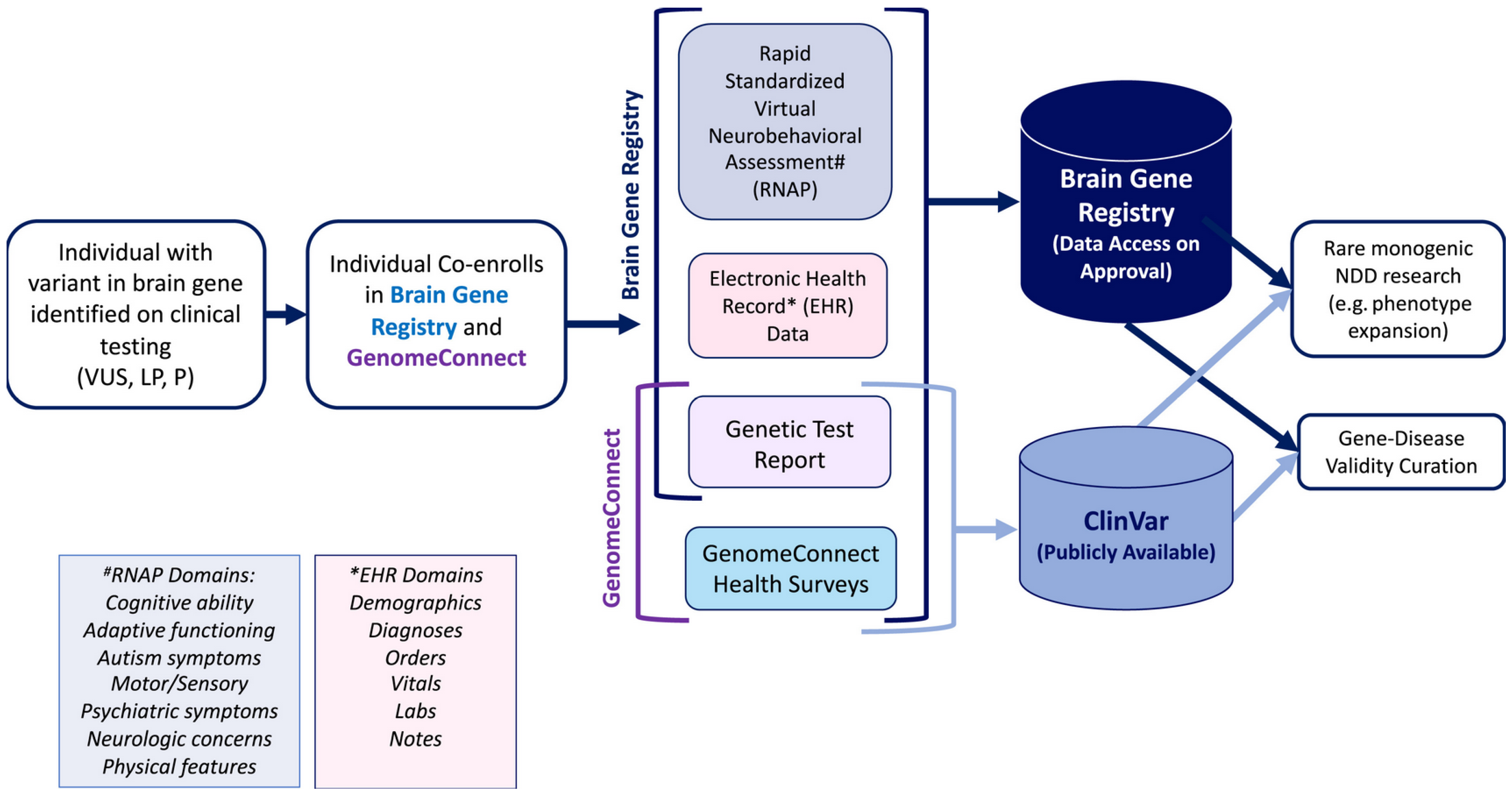
2. Brain Gene Registry
2.1. Background
2.2. Expanding Current Databases
2.3. How the BGR Will Inform Clinical Trial Design
2.4. Site Enrollment, Data Security & Patient Privacy
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NDD | Neurodevelopmental Disorder |
| ID | Intellectual Disability |
| BGR | Brain Gene Registry |
References
- Jeyabalan, N.; Clement, J.P. SYNGAP1: Mind the Gap. Front. Cell. Neurosci. 2016, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Hubel, D.H. The Brain. Sci. Am. 1979, 241, 44–53. [Google Scholar] [CrossRef]
- Agarwal, M.; Johnston, M.V.; Stafstrom, C.E. SYNGAP1 mutations: Clinical, genetic, and pathophysiological features. Int. J. Dev. Neurosci. 2019, 78, 65–76. [Google Scholar] [CrossRef]
- Chen, H.-J.; Rojas-Soto, M.; Oguni, A.; Kennedy, M.B. A synaptic Ras-GTPase activating protein (p135 SynGAP) inhibited by CaM kinase II. Neuron 1998, 20, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Liao, D.; Lau, L.-F.; Huganir, R.L. SynGAP: A Synaptic RasGAP that associates with the PSD-95/SAP90 protein family. Neuron 1998, 20, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Gamache, T.R.; Araki, Y.; Huganir, R.L. Twenty Years of SynGAP Research: From Synapses to Cognition. J. Neurosci. 2020, 40, 1596–1605. [Google Scholar] [CrossRef]
- Araki, Y.; Rajkovich, K.E.; Gerber, E.E.; Gamache, T.R.; Johnson, R.C.; Tran, T.H.N.; Liu, B.; Zhu, Q.; Hong, I.; Kirkwood, A.; et al. SynGAP regulates synaptic plasticity and cognition independently of its catalytic activity. Science 2024, 383, eadk1291. [Google Scholar] [CrossRef]
- Baldridge, D.; Kaster, L.; Sancimino, C.; Srivastava, S.; Molholm, S.; Gupta, A.; Oh, I.; Lanzotti, V.; Grewal, D.; Riggs, E.R.; et al. The Brain Gene Registry: A data snapshot. J. Neurodev. Disord. 2024, 16, 17. [Google Scholar] [CrossRef]
- Chopra, M.; Savatt, J.M.; Bingaman, T.I.; Good, M.E.; Morgan, A.; Cooney, C.; Rossel, A.M.; VanHoute, B.; Cordova, I.; Mahida, S.; et al. Clinical variants paired with phenotype: A rich resource for brain gene curation. Anesth. Analg. 2024, 26, 101035. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS proteins and their regulators in human disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Gauthier, J.; Spiegelman, D.; Noreau, A.; Yang, Y.; Pellerin, S.; Dobrzeniecka, S.; Côté, M.; Perreau-Linck, E.; Carmant, L.; et al. Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N. Engl. J. Med. 2009, 360, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Holder, J.L.; Jr Hamdan, F.F.; Michaud, J.L. SYNGAP1-Related Intellectual Disability. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. Available online: https://www.ncbi.nlm.nih.gov/pubmed/30789692 (accessed on 15 January 2025).
- Weldon, M.; Kilinc, M.; Holder, J.L., Jr.; Rumbaugh, G. The first international conference on SYNGAP1-related brain disorders: A stakeholder meeting of families, researchers, clinicians, and regulators. J. Neurodev. Disord. 2018, 10, 6. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, H.; Yang, S.; Li, J.; Xie, H.; Chen, X. Correlation of early neurodevelopmental features of children with SYNGAP1 variants and their genotypes. Zhonghua Yi Xue Yi Chuan Xue Za Zhi = Zhonghua Yixue Yichuanxue Zazhi = Chin. J. Med. Genet. 2024, 41, 25–31. (In Chinese) [Google Scholar] [CrossRef] [PubMed]
- Mignot, C.; von Stülpnagel, C.; Nava, C.; Ville, D.; Sanlaville, D.; Lesca, G.; Rastetter, A.; Gachet, B.; Marie, Y.; Korenke, G.C.; et al. Genetic and neurodevelopmental spectrum of SYNGAP1-associated intellectual disability and epilepsy. J. Med Genet. 2016, 53, 511–522, Erratum in J. Med. Genet. 2016, 53, 720. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Gomez, A.; Niu, S.; Andujar-Perez, F.; McQuade, E.A.; Balasa, A.; Huss, D.; Coorg, R.; Quach, M.; Vinson, S.; Risen, S.; et al. Phenotypic characterization of individuals with SYNGAP1 pathogenic variants reveals a potential correlation between posterior dominant rhythm and developmental progression. J. Neurodev. Disord. 2019, 11, 18. [Google Scholar] [CrossRef]
- Carvill, G.L.; Heavin, S.B.; Yendle, S.C.; McMahon, J.M.; O’Roak, B.J.; Cook, J.; Khan, A.; Dorschner, M.O.; Weaver, M.; Calvert, S.; et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat. Genet. 2013, 45, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Wiltrout, K.; Brimble, E.; Poduri, A. Comprehensive phenotypes of patients with SYNGAP1-related disorder reveals high rates of epilepsy and autism. Epilepsia 2024, 65, 1428–1438. [Google Scholar] [CrossRef]
- Wright, D.; Kenny, A.; Eley, S.; McKechanie, A.G.; Stanfield, A.C. Clinical and behavioural features of SYNGAP1-related intellectual disability: A parent and caregiver description. J. Neurodev. Disord. 2022, 14, 34. [Google Scholar] [CrossRef]
- Michaelson, S.D.; Ozkan, E.D.; Aceti, M.; Maity, S.; Llamosas, N.; Weldon, M.; Mizrachi, E.; Vaissiere, T.; Gaffield, M.A.; Christie, J.M.; et al. SYNGAP1 heterozygosity disrupts sensory processing by reducing touch-related activity within somatosensory cortex circuits. Nat. Neurosci. 2018, 21, 1–13. [Google Scholar] [CrossRef]
- Baldwin, I.; Cho, A.; Orenstein, G.; Wilner, N.; Nicoli, D.; Smith, J.R. SYNGAP-1 Mutation And Catatonia: A Case Series and Systematic Review. J. Child Adolesc. Psychopharmacol. 2024, 34, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Rong, M.; Benke, T.; Ali, Q.Z.; Aledo-Serrano, Á.; Bayat, A.; Rossi, A.; Devinsky, O.; Qaiser, F.; Ali, A.S.; Fasano, A.; et al. Adult Phenotype of SYNGAP1-DEE. Neurol. Genet. 2023, 9, e200105. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Greco, M.; Uittenbogaard, M.; Chiaramello, A.E.; Gropman, A.L. The unexpected and novel mitochondrial phenotype of the ex vivo patient-derived cellular model for SYNGAP1 encephalopathy [CNS Annual Meeting abstract PL2-6]. Ann. Neurol. 2024, 96, S1–S162. [Google Scholar]
- Han, F.Y.; Conboy-Schmidt, L.; Rybachuk, G.; Volk, H.A.; Zanghi, B.; Pan, Y.; Borges, K. Dietary medium chain triglycerides for management of epilepsy: New data from human, dog, and rodent studies. Epilepsia 2021, 62, 1790–1806. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dawicki-McKenna, J.M.; Felix, A.J.; Waxman, E.A.; Cheng, C.; Amado, D.A.; Ranum, P.T.; Bogush, A.; Dungan, L.V.; Maguire, J.A.; Gagne, A.L.; et al. Mapping PTBP2 binding in human brain identifies SYNGAP1 as a target for therapeutic splice switching. Nat. Commun. 2023, 14, 2628. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Hwang, Y.; Ludwig, N.; Henry, J.; Kadam, S.D. Case report: Off-label use of low-dose perampanel in a 25-month-old girl with a pathogenic SYNGAP1 variant. Front. Neurol. 2023, 14, 1221161. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Syngap Research Fund. Cure Syngap1. 4 January 2025. Available online: https://curesyngap1.org/blog/syngap-research-funds-2024-syngap1-science-conference-collaboration-and-progress-on-our-road-to-cure-syngap1/ (accessed on 7 February 2025).
- Halley, M.C.; Smith, H.S.; Ashley, E.A.; Goldenberg, A.J.; Tabor, H.K. A call for an integrated approach to improve efficiency, equity and sustainability in rare disease research in the United States. Nat. Genet. 2022, 54, 219–222. [Google Scholar] [CrossRef]
- Cordova, I.; Blesson, A.; Savatt, J.M.; Sveden, A.; Mahida, S.; Hazlett, H.; Riggs, E.R.; Chopra, M. Brain Gene Registry Subset of the ClinGen Intellectual Disability and Autism Gene Curation Expert Panel. Expansion of the Genotypic and Phenotypic Spectrum of ASH1L-Related Syndromic Neurodevelopmental Disorder. Genes 2024, 15, 423. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sara Dick, M. CCRP: Minimally Disruptive Research: A Respectful Approach to Conducting Clinical Studies; UNIT K, Ed.; The Knowledge and Evaluation Research Unit at Mayo Clinic: Rochester, MN, USA, 2017. [Google Scholar]
- Screening Consent. National Brain Gene Registry. Available online: https://braingeneregistry.wustl.edu/screening-consent/ (accessed on 22 March 2025).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greco, M.R.; Chatterjee, M.; Taylor, A.M.; Gropman, A.L. SYNGAP1 Syndrome and the Brain Gene Registry. Genes 2025, 16, 405. https://doi.org/10.3390/genes16040405
Greco MR, Chatterjee M, Taylor AM, Gropman AL. SYNGAP1 Syndrome and the Brain Gene Registry. Genes. 2025; 16(4):405. https://doi.org/10.3390/genes16040405
Chicago/Turabian StyleGreco, Melissa R., Maya Chatterjee, Alexa M. Taylor, and Andrea L. Gropman. 2025. "SYNGAP1 Syndrome and the Brain Gene Registry" Genes 16, no. 4: 405. https://doi.org/10.3390/genes16040405
APA StyleGreco, M. R., Chatterjee, M., Taylor, A. M., & Gropman, A. L. (2025). SYNGAP1 Syndrome and the Brain Gene Registry. Genes, 16(4), 405. https://doi.org/10.3390/genes16040405