
Molecular Landscape and Diagnostic Model of MASH: Transcriptomic, Proteomic, Metabolomic, and Lipidomic Perspectives



Abstract
1. Introduction
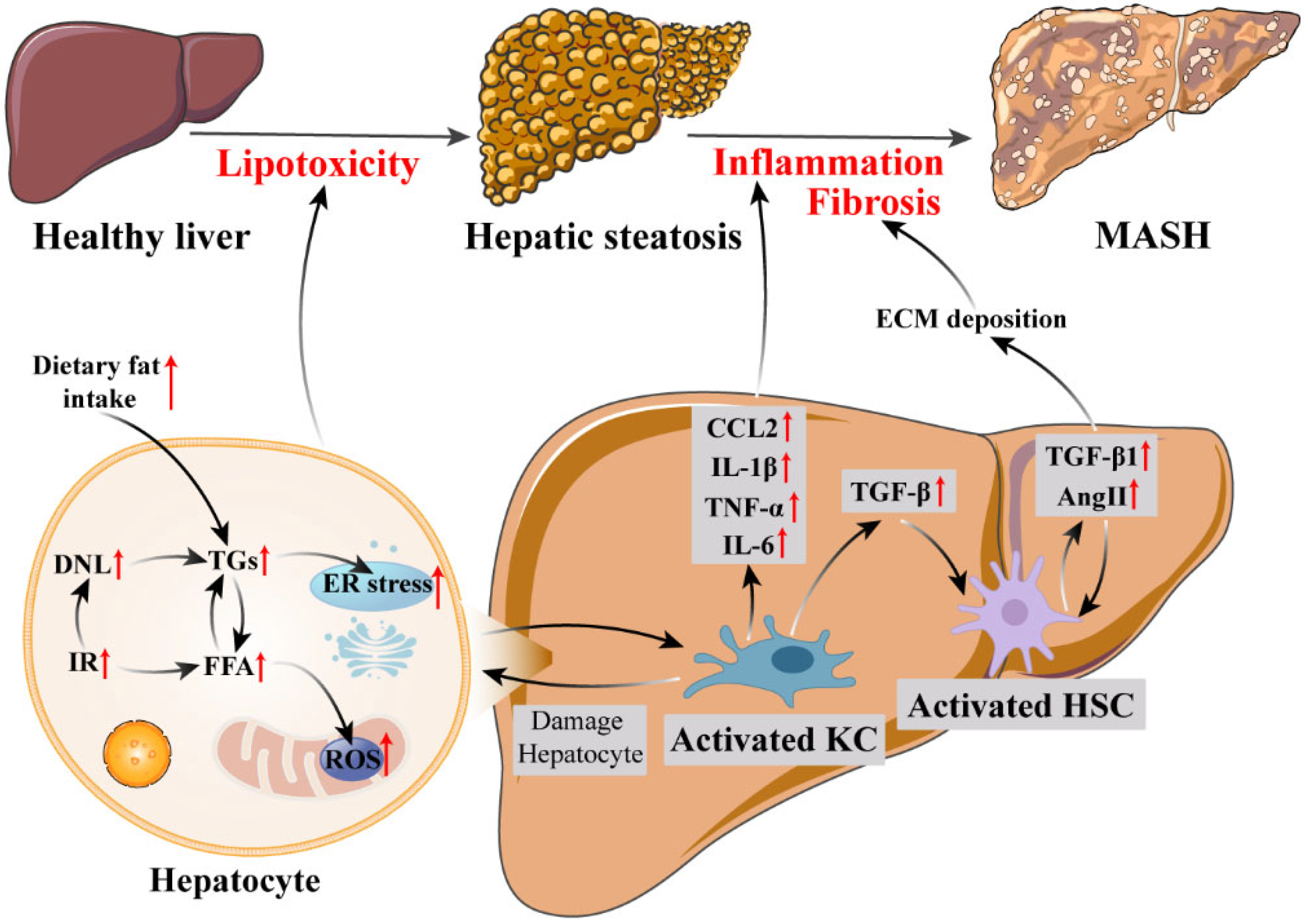
2. Pathophysiology of MASH
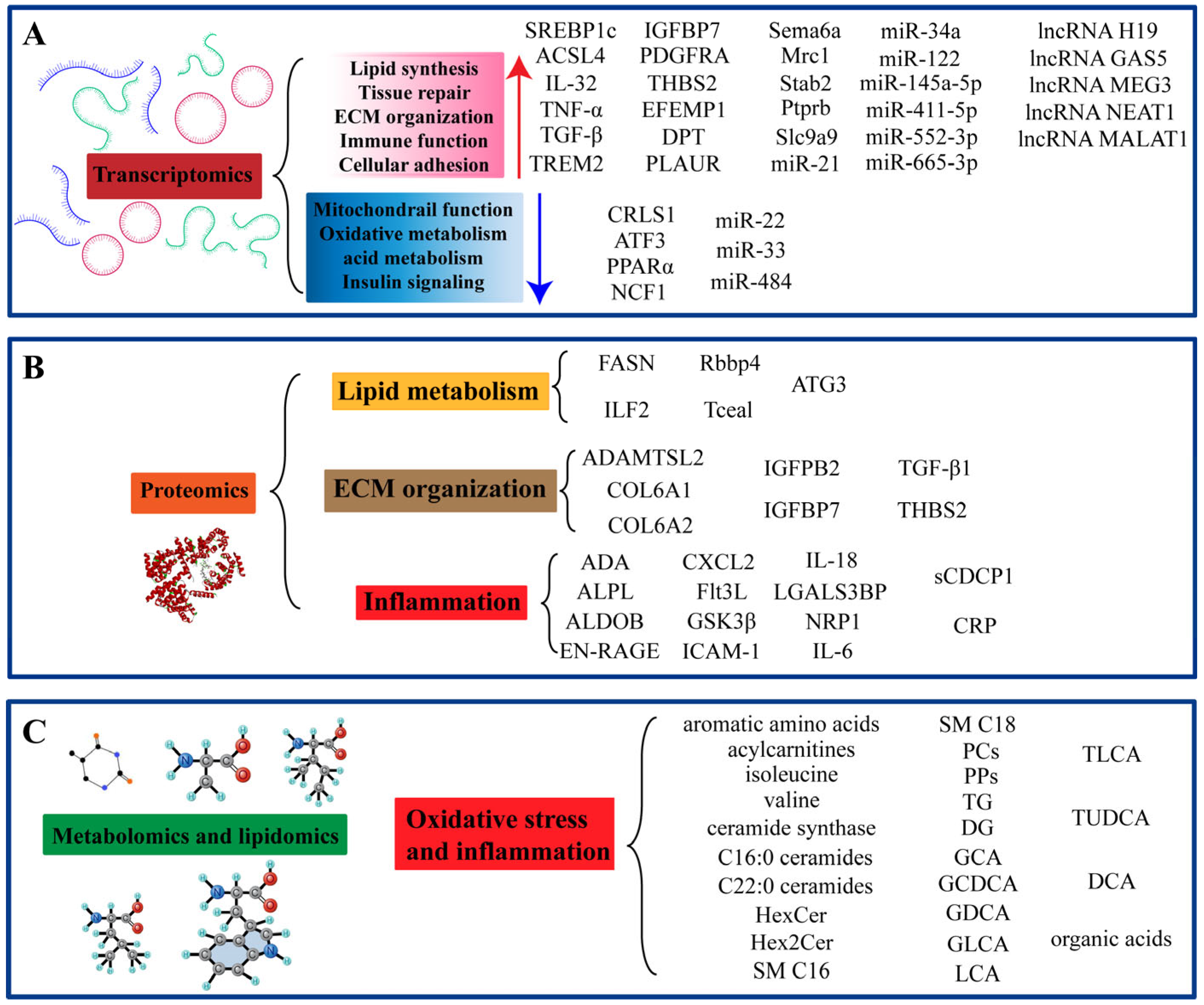
3. Transcriptomics
3.1. Gene Expression and Transcriptome Profiling Analysis
3.2. MicroRNAs and Long Non-Coding RNAs
3.3. Diagnostic Model Based on Transcriptomics
4. Proteomics
4.1. Protein Expression and Protein Profiling Analysis
4.2. Diagnostic Model Based on Proteomics
5. Metabolomics and Lipidomics
5.1. The Role of Lipid and Metabolite Remodeling in MASH/MASLD Pathogenesis
5.2. Diagnostic Models Based on Metabolomics and Lipidomics
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. J. Hepatol. 2023, 79, 1542–1556. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; Price, J.K.; Owrangi, S.; Gundu-Rao, N.; Satchi, R.; Paik, J.M. The Global Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis Among Patients With Type 2 Diabetes. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2024, 22, 1999–2010.e8. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Kalligeros, M.; Henry, L. Epidemiology of Metabolic Dysfunction-Associated Steatotic Liver Disease. Clin. Mol. Hepatol. 2024, 31, S32–S50. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; Paik, J.; Owrangi, S.; Yilmaz, Y.; El-Kassas, M.; Alswat, K.; Alqahtani, S.A. Prevalence of metabolic dysfunction-associated steatotic liver disease in the Middle East and North Africa. Liver Int. Off. J. Int. Assoc. Study Liver 2024, 44, 1061–1070. [Google Scholar] [CrossRef]
- Tulone, A.; Pennisi, G.; Ciccioli, C.; Infantino, G.; La Mantia, C.; Cannella, R.; Mercurio, F.; Petta, S. Are we ready for genetic testing in metabolic dysfunction-associated steatotic liver disease? United Eur. Gastroenterol. J. 2024, 12, 638–648. [Google Scholar] [CrossRef]
- Cathcart, J.; Barrett, R.; Bowness, J.S.; Mukhopadhya, A.; Lynch, R.; Dillon, J.F. Accuracy of Non-Invasive Imaging Techniques for the Diagnosis of MASH in Patients with MASLD: A Systematic Review. Liver Int. Off. J. Int. Assoc. Study Liver 2024, 45, e16127. [Google Scholar] [CrossRef]
- Chan, W.K.; Wong, V.W.; Adams, L.A.; Nguyen, M.H. MAFLD in adults: Non-invasive tests for diagnosis and monitoring of MAFLD. Hepatol. Int. 2024, 18, 909–921. [Google Scholar] [CrossRef]
- Chandra Kumar, C.V.; Skantha, R.; Chan, W.K. Non-invasive assessment of metabolic dysfunction-associated fatty liver disease. Ther. Adv. Endocrinol. Metab. 2022, 13, 20420188221139614. [Google Scholar] [CrossRef]
- Hayes, C.N.; Nakahara, H.; Ono, A.; Tsuge, M.; Oka, S. From Omics to Multi-Omics: A Review of Advantages and Tradeoffs. Genes 2024, 15, 1551. [Google Scholar] [CrossRef]
- Reel, P.S.; Reel, S.; Pearson, E.; Trucco, E.; Jefferson, E. Using machine learning approaches for multi-omics data analysis: A review. Biotechnol. Adv. 2021, 49, 107739. [Google Scholar] [CrossRef]
- Perakakis, N.; Stefanakis, K.; Mantzoros, C.S. The role of omics in the pathophysiology, diagnosis and treatment of non-alcoholic fatty liver disease. Metab. Clin. Exp. 2020, 111, 154320. [Google Scholar] [CrossRef]
- Long, Q.; Luo, F.; Li, B.; Li, Z.; Guo, Z.; Chen, Z.; Wu, W.; Hu, M. Gut microbiota and metabolic biomarkers in metabolic dysfunction-associated steatotic liver disease. Hepatol. Commun. 2024, 8, e0310. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, A.; Gentilini, A.; Marra, F. Molecular Pathogenesis of NASH. Int. J. Mol. Sci. 2016, 17, 1575. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef]
- Fujita, K.; Nozaki, Y.; Wada, K.; Yoneda, M.; Fujimoto, Y.; Fujitake, M.; Endo, H.; Takahashi, H.; Inamori, M.; Kobayashi, N.; et al. Dysfunctional very-low-density lipoprotein synthesis and release is a key factor in nonalcoholic steatohepatitis pathogenesis. Hepatology 2009, 50, 772–780. [Google Scholar] [CrossRef]
- Zhang, X.Q.; Xu, C.F.; Yu, C.H.; Chen, W.X.; Li, Y.M. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 1768–1776. [Google Scholar] [CrossRef]
- Ashraf, N.U.; Sheikh, T.A. Endoplasmic reticulum stress and Oxidative stress in the pathogenesis of Non-alcoholic fatty liver disease. Free Radic. Res. 2015, 49, 1405–1418. [Google Scholar] [CrossRef]
- Wenfeng, Z.; Yakun, W.; Di, M.; Jianping, G.; Chuanxin, W.; Chun, H. Kupffer cells: Increasingly significant role in nonalcoholic fatty liver disease. Ann. Hepatol. 2014, 13, 489–495. [Google Scholar]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef]
- Rolo, A.P.; Teodoro, J.S.; Palmeira, C.M. Role of oxidative stress in the pathogenesis of nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2012, 52, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Puche, J.E.; Saiman, Y.; Friedman, S.L. Hepatic stellate cells and liver fibrosis. Compr. Physiol. 2013, 3, 1473–1492. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; ten Dijke, P. TGF-β in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Rajapaksha, I.G.; Gunarathne, L.S.; Angus, P.W.; Herath, C.B. Update on New Aspects of the Renin-Angiotensin System in Hepatic Fibrosis and Portal Hypertension: Implications for Novel Therapeutic Options. J. Clin. Med. 2021, 10, 702. [Google Scholar] [CrossRef]
- Moreira de Macêdo, S.; Guimarães, T.A.; Feltenberger, J.D.; Sousa Santos, S.H. The role of renin-angiotensin system modulation on treatment and prevention of liver diseases. Peptides 2014, 62, 189–196. [Google Scholar] [CrossRef]
- Stahl, F.; Hitzmann, B.; Mutz, K.; Landgrebe, D.; Lübbecke, M.; Kasper, C.; Walter, J.; Scheper, T. Transcriptome analysis. Adv. Biochem. Eng. Biotechnol. 2012, 127, 1–25. [Google Scholar] [CrossRef]
- Lee, E.H.; Lee, J.H.; Kim, D.Y.; Lee, Y.S.; Jo, Y.; Dao, T.; Kim, K.E.; Song, D.K.; Seo, J.H.; Seo, Y.K.; et al. Loss of SREBP-1c ameliorates iron-induced liver fibrosis by decreasing lipocalin-2. Exp. Mol. Med. 2024, 56, 1001–1012. [Google Scholar] [CrossRef]
- Tang, K.; Zhang, H.; Deng, J.; Wang, D.; Liu, S.; Lu, S.; Cui, Q.; Chen, C.; Liu, J.; Yang, Z.; et al. Ammonia detoxification promotes CD8(+) T cell memory development by urea and citrulline cycles. Nat. Immunol. 2023, 24, 162–173. [Google Scholar] [CrossRef]
- Baselli, G.A.; Dongiovanni, P.; Rametta, R.; Meroni, M.; Pelusi, S.; Maggioni, M.; Badiali, S.; Pingitore, P.; Maurotti, S.; Montalcini, T.; et al. Liver transcriptomics highlights interleukin-32 as novel NAFLD-related cytokine and candidate biomarker. Gut 2020, 69, 1855–1866. [Google Scholar] [CrossRef]
- Tu, C.; Xiong, H.; Hu, Y.; Wang, W.; Mei, G.; Wang, H.; Li, Y.; Zhou, Z.; Meng, F.; Zhang, P.; et al. Cardiolipin Synthase 1 Ameliorates NASH Through Activating Transcription Factor 3 Transcriptional Inactivation. Hepatology 2020, 72, 1949–1967. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.; Pouryahya, M.; Gindin, Y.; Pan, D.Z.; Carrasco-Zevallos, O.M.; Mountain, V.; Subramanian, G.M.; Montalto, M.C.; Resnick, M.; Beck, A.H.; et al. Integration of deep learning-based histopathology and transcriptomics reveals key genes associated with fibrogenesis in patients with advanced NASH. Cell Reports. Med. 2023, 4, 101016. [Google Scholar] [CrossRef]
- Moylan, C.A.; Pang, H.; Dellinger, A.; Suzuki, A.; Garrett, M.E.; Guy, C.D.; Murphy, S.K.; Ashley-Koch, A.E.; Choi, S.S.; Michelotti, G.A.; et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014, 59, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Sun, C.; Hou, Y.; Tang, Y.; Zhu, Z.; Zhang, Z.; Zhang, Y.; Wang, L.; Zhao, Q.; Chen, M.G.; et al. A comprehensive bioinformatics analysis on multiple Gene Expression Omnibus datasets of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Sci. Rep. 2018, 8, 7630. [Google Scholar] [CrossRef]
- Kozumi, K.; Kodama, T.; Murai, H.; Sakane, S.; Govaere, O.; Cockell, S.; Motooka, D.; Kakita, N.; Yamada, Y.; Kondo, Y.; et al. Transcriptomics Identify Thrombospondin-2 as a Biomarker for NASH and Advanced Liver Fibrosis. Hepatology 2021, 74, 2452–2466. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Gorreta, F.; Ong, J.P.; Schlauch, K.; Del Giacco, L.; Elariny, H.; Van Meter, A.; Younoszai, A.; Goodman, Z.; Baranova, A.; et al. Hepatic gene expression in patients with obesity-related non-alcoholic steatohepatitis. Liver Int. 2005, 25, 760–771. [Google Scholar] [CrossRef]
- Duan, J.; Wang, Z.; Duan, R.; Yang, C.; Zhao, R.; Feng, Q.; Qin, Y.; Jiang, J.; Gu, S.; Lv, K.; et al. Therapeutic targeting of hepatic ACSL4 ameliorates NASH in mice. Hepatology 2022, 75, 140–153. [Google Scholar] [CrossRef]
- Lefebvre, P.; Lalloyer, F.; Baugé, E.; Pawlak, M.; Gheeraert, C.; Dehondt, H.; Vanhoutte, J.; Woitrain, E.; Hennuyer, N.; Mazuy, C.; et al. Interspecies NASH disease activity whole-genome profiling identifies a fibrogenic role of PPARα-regulated dermatopontin. JCI Insight 2017, 2, e92264. [Google Scholar] [CrossRef]
- Yashaswini, C.N.; Qin, T.; Bhattacharya, D.; Amor, C.; Lowe, S.; Lujambio, A.; Wang, S.; Friedman, S.L. Phenotypes and ontogeny of senescent hepatic stellate cells in metabolic dysfunction-associated steatohepatitis. J. Hepatol. 2024, 81, 207–217. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Fan, M.; Guan, Y.; Zhang, W.; Huang, F.; Zhang, Z.; Li, X.; Yuan, B.; Liu, W.; et al. Reactive oxygen species regulation by NCF1 governs ferroptosis susceptibility of Kupffer cells to MASH. Cell Metab. 2024, 36, 1745–1763.e6. [Google Scholar] [CrossRef]
- Bendixen, S.M.; Jakobsgaard, P.R.; Hansen, D.; Hejn, K.H.; Terkelsen, M.K.; Bjerre, F.A.; Thulesen, A.P.; Eriksen, N.G.; Hallenborg, P.; Geng, Y.; et al. Single cell-resolved study of advanced murine MASH reveals a homeostatic pericyte signaling module. J. Hepatol. 2024, 80, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.Y.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol. Cell 2019, 75, 644–660.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Lu, Y.; Qiu, X.; Chen, Z.; Tang, Y.; Meng, Z.; Yan, C.; Du, H.; Li, S.; Lin, J.D. Lipid droplet efferocytosis attenuates proinflammatory signaling in macrophages via TREM2- and MS4A7-dependent mechanisms. Cell Rep. 2025, 44, 115310. [Google Scholar] [CrossRef] [PubMed]
- Hendrikx, T.; Porsch, F.; Kiss, M.G.; Rajcic, D.; Papac-Miličević, N.; Hoebinger, C.; Goederle, L.; Hladik, A.; Shaw, L.E.; Horstmann, H.; et al. Soluble TREM2 levels reflect the recruitment and expansion of TREM2+ macrophages that localize to fibrotic areas and limit NASH. J. Hepatol. 2022, 77, 1373–1385. [Google Scholar] [CrossRef]
- Zhou, L.; Qiu, X.; Meng, Z.; Liu, T.; Chen, Z.; Zhang, P.; Kuang, H.; Pan, T.; Lu, Y.; Qi, L.; et al. Hepatic danger signaling triggers TREM2+ macrophage induction and drives steatohepatitis via MS4A7-dependent inflammasome activation. Sci. Transl. Med. 2024, 16, eadk1866. [Google Scholar] [CrossRef]
- Rosenthal, S.B.; Liu, X.; Ganguly, S.; Dhar, D.; Pasillas, M.P.; Ricciardelli, E.; Li, R.Z.; Troutman, T.D.; Kisseleva, T.; Glass, C.K.; et al. Heterogeneity of HSCs in a Mouse Model of NASH. Hepatology 2021, 74, 667–685. [Google Scholar] [CrossRef]
- Payen, V.L.; Lavergne, A.; Alevra Sarika, N.; Colonval, M.; Karim, L.; Deckers, M.; Najimi, M.; Coppieters, W.; Charloteaux, B.; Sokal, E.M.; et al. Single-cell RNA sequencing of human liver reveals hepatic stellate cell heterogeneity. JHEP Rep. Innov. Hepatol. 2021, 3, 100278. [Google Scholar] [CrossRef]
- Breuer, D.A.; Pacheco, M.C.; Washington, M.K.; Montgomery, S.A.; Hasty, A.H.; Kennedy, A.J. CD8+ T cells regulate liver injury in obesity-related nonalcoholic fatty liver disease. Am. J. Physiology. Gastrointest. Liver Physiol. 2020, 318, G211–G224. [Google Scholar] [CrossRef]
- Carter, J.K.; Friedman, S.L. Hepatic Stellate Cell-Immune Interactions in NASH. Front. Endocrinol. 2022, 13, 867940. [Google Scholar] [CrossRef]
- Gribben, C.; Galanakis, V.; Calderwood, A.; Williams, E.C.; Chazarra-Gil, R.; Larraz, M.; Frau, C.; Puengel, T.; Guillot, A.; Rouhani, F.J.; et al. Acquisition of epithelial plasticity in human chronic liver disease. Nature 2024, 630, 166–173. [Google Scholar] [CrossRef]
- Jin, X.; Gao, J.; Zheng, R.; Yu, M.; Ren, Y.; Yan, T.; Huang, Y.; Li, Y. Antagonizing circRNA_002581-miR-122-CPEB1 axis alleviates NASH through restoring PTEN-AMPK-mTOR pathway regulated autophagy. Cell Death Dis. 2020, 11, 123. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.T.; Yang, M.Y.; Lee, Y.J.; Yang, T.W.; Wang, C.C.; Wang, C.J. Gallic Acid Improves Diabetic Steatosis by Downregulating MicroRNA-34a-5p through Targeting NFE2L2 Expression in High-Fat Diet-Fed db/db Mice. Antioxidants 2021, 11, 92. [Google Scholar] [CrossRef] [PubMed]
- Long, J.K.; Dai, W.; Zheng, Y.W.; Zhao, S.P. miR-122 promotes hepatic lipogenesis via inhibiting the LKB1/AMPK pathway by targeting Sirt1 in non-alcoholic fatty liver disease. Mol. Med. 2019, 25, 26. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Tussy, P.; Cardelo, M.P.; Zhang, H.; Sun, J.; Price, N.L.; Boutagy, N.E.; Goedeke, L.; Cadena-Sandoval, M.; Xirouchaki, C.E.; Brown, W.; et al. miR-33 deletion in hepatocytes attenuates MASLD-MASH-HCC progression. JCI Insight 2024, 9, e168476. [Google Scholar] [CrossRef]
- Panella, R.; Petri, A.; Desai, B.N.; Fagoonee, S.; Cotton, C.A.; Nguyen, P.K.; Lundin, E.M.; Wagshal, A.; Wang, D.Z.; Näär, A.M.; et al. MicroRNA-22 Is a Key Regulator of Lipid and Metabolic Homeostasis. Int. J. Mol. Sci. 2023, 24, 12870. [Google Scholar] [CrossRef]
- Jia, Y.; Pan, H.; Liu, J.; Huang, T.; Han, K.; Mei, Q.; Zeng, L.; Zhou, J.; Zhang, Y. MiR-484 promotes nonalcoholic fatty liver disease progression in mice via downregulation of Sorbs2. Obesity 2023, 31, 2972–2985. [Google Scholar] [CrossRef]
- Ezaz, G.; Trivedi, H.D.; Connelly, M.A.; Filozof, C.; Howard, K.; Parrish, M.L.; Kim, M.; Herman, M.A.; Nasser, I.; Afdhal, N.H.; et al. Differential Associations of Circulating MicroRNAs With Pathogenic Factors in NAFLD. Hepatol. Commun. 2020, 4, 670–680. [Google Scholar] [CrossRef]
- Pirola, C.J.; Fernández Gianotti, T.; Castaño, G.O.; Mallardi, P.; San Martino, J.; Mora Gonzalez Lopez Ledesma, M.; Flichman, D.; Mirshahi, F.; Sanyal, A.J.; Sookoian, S. Circulating microRNA signature in non-alcoholic fatty liver disease: From serum non-coding RNAs to liver histology and disease pathogenesis. Gut 2015, 64, 800–812. [Google Scholar] [CrossRef]
- Cheung, O.; Puri, P.; Eicken, C.; Contos, M.J.; Mirshahi, F.; Maher, J.W.; Kellum, J.M.; Min, H.; Luketic, V.A.; Sanyal, A.J. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology 2008, 48, 1810–1820. [Google Scholar] [CrossRef]
- Li, X.; Zhang, W.; Xu, K.; Lu, J. miR-34a promotes liver fibrosis in patients with chronic hepatitis via mediating Sirt1/p53 signaling pathway. Pathol. Res. Pract. 2020, 216, 152876. [Google Scholar] [CrossRef]
- Lin, Y.; Ding, D.; Huang, Q.; Liu, Q.; Lu, H.; Lu, Y.; Chi, Y.; Sun, X.; Ye, G.; Zhu, H.; et al. Downregulation of miR-192 causes hepatic steatosis and lipid accumulation by inducing SREBF1: Novel mechanism for bisphenol A-triggered non-alcoholic fatty liver disease. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2017, 1862, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Huang, F.; Liu, X.; Xiao, X.; Yang, M.; Hu, G.; Liu, H.; Liao, L. miR-21 regulates triglyceride and cholesterol metabolism in non-alcoholic fatty liver disease by targeting HMGCR. Int. J. Mol. Med. 2015, 35, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.M.; Afonso, M.B.; Simão, A.L.; Carvalho, C.C.; Trindade, A.; Duarte, A.; Borralho, P.M.; Machado, M.V.; Cortez-Pinto, H.; Rodrigues, C.M.; et al. miR-21 ablation and obeticholic acid ameliorate nonalcoholic steatohepatitis in mice. Cell Death Dis. 2017, 8, e2748. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yang, Z.; Mao, F.; Gong, W.; Su, Q.; Yang, J.; Liu, B.; Song, Y.; Jin, J.; Lu, Y. Downregulation of microRNA-145a-5p promotes steatosis-to-NASH progression through upregulation of Nr4a2. J. Hepatol. 2023, 79, 1096–1109. [Google Scholar] [CrossRef]
- Ma, N.; Hou, A.; Pan, X.; Sun, F.; Xu, X.; Yu, C.; Lai, R.; Huang, R.; Gong, L.; Xie, Q.; et al. MiR-552-3p Regulates Multiple Fibrotic and Inflammatory genes Concurrently in Hepatic Stellate Cells Improving NASH-associated Phenotypes. Int. J. Biol. Sci. 2023, 19, 3456–3471. [Google Scholar] [CrossRef]
- Yu, Y.; Tian, T.; Tan, S.; Wu, P.; Guo, Y.; Li, M.; Huang, M. MicroRNA-665-3p exacerbates nonalcoholic fatty liver disease in mice. Bioengineered 2022, 13, 2927–2942. [Google Scholar] [CrossRef]
- Wan, Z.; Yang, X.; Liu, X.; Sun, Y.; Yu, P.; Xu, F.; Deng, H. M2 macrophage-derived exosomal microRNA-411-5p impedes the activation of hepatic stellate cells by targeting CAMSAP1 in NASH model. iScience 2022, 25, 104597. [Google Scholar] [CrossRef]
- Fu, X.; Zhu, J.; Zhang, L.; Shu, J. Long non-coding RNA NEAT1 promotes steatosis via enhancement of estrogen receptor alpha-mediated AQP7 expression in HepG2 cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1782–1787. [Google Scholar] [CrossRef]
- Wang, X. Down-regulation of lncRNA-NEAT1 alleviated the non-alcoholic fatty liver disease via mTOR/S6K1 signaling pathway. J. Cell. Biochem. 2018, 119, 1567–1574. [Google Scholar] [CrossRef]
- Jin, S.S.; Lin, C.J.; Lin, X.F.; Zheng, J.Z.; Guan, H.Q. Silencing lncRNA NEAT1 reduces nonalcoholic fatty liver fat deposition by regulating the miR-139-5p/c-Jun/SREBP-1c pathway. Ann. Hepatol. 2022, 27, 100584. [Google Scholar] [CrossRef]
- Xiang, J.; Deng, Y.Y.; Liu, H.X.; Pu, Y. LncRNA MALAT1 Promotes PPARα/CD36-Mediated Hepatic Lipogenesis in Nonalcoholic Fatty Liver Disease by Modulating miR-206/ARNT Axis. Front. Bioeng. Biotechnol. 2022, 10, 858558. [Google Scholar] [CrossRef]
- Liu, C.; Yang, Z.; Wu, J.; Zhang, L.; Lee, S.; Shin, D.J.; Tran, M.; Wang, L. Long noncoding RNA H19 interacts with polypyrimidine tract-binding protein 1 to reprogram hepatic lipid homeostasis. Hepatology 2018, 67, 1768–1783. [Google Scholar] [CrossRef] [PubMed]
- Zou, D.; Liu, L.; Zeng, Y.; Wang, H.; Dai, D.; Xu, M. LncRNA MEG3 up-regulates SIRT6 by ubiquitinating EZH2 and alleviates nonalcoholic fatty liver disease. Cell Death Discov. 2022, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Huang, F.Z.; Liu, H.Z.; Zhang, T.Y.; Yang, M.S.; Sun, C.Z. LncRNA MEG3 functions as a ceRNA in regulating hepatic lipogenesis by competitively binding to miR-21 with LRP6. Metab. Clin. Exp. 2019, 94, 1–8. [Google Scholar] [CrossRef]
- Erdem, M.G.; Unlu, O.; Demirci, M. Could Long Non-Coding RNA MEG3 and PTENP1 Interact with miR-21 in the Pathogenesis of Non-Alcoholic Fatty Liver Disease? Biomedicines 2023, 11, 574. [Google Scholar] [CrossRef]
- Di Mauro, S.; Scamporrino, A.; Petta, S.; Urbano, F.; Filippello, A.; Ragusa, M.; Di Martino, M.T.; Scionti, F.; Grimaudo, S.; Pipitone, R.M.; et al. Serum coding and non-coding RNAs as biomarkers of NAFLD and fibrosis severity. Liver Int. Off. J. Int. Assoc. Study Liver 2019, 39, 1742–1754. [Google Scholar] [CrossRef]
- Cui, J.; Wang, Y.; Xue, H. Long non-coding RNA GAS5 contributes to the progression of nonalcoholic fatty liver disease by targeting the microRNA-29a-3p/NOTCH2 axis. Bioengineered 2022, 13, 8370–8381. [Google Scholar] [CrossRef]
- Newsome, P.N.; Sasso, M.; Deeks, J.J.; Paredes, A.; Boursier, J.; Chan, W.K.; Yilmaz, Y.; Czernichow, S.; Zheng, M.H.; Wong, V.W.; et al. FibroScan-AST (FAST) score for the non-invasive identification of patients with non-alcoholic steatohepatitis with significant activity and fibrosis: A prospective derivation and global validation study. Lancet. Gastroenterol. Hepatol. 2020, 5, 362–373. [Google Scholar] [CrossRef]
- Kjaergaard, M.; Lindvig, K.P.; Thorhauge, K.H.; Andersen, P.; Hansen, J.K.; Kastrup, N.; Jensen, J.M.; Hansen, C.D.; Johansen, S.; Israelsen, M.; et al. Using the ELF test, FIB-4 and NAFLD fibrosis score to screen the population for liver disease. J. Hepatol. 2023, 79, 277–286. [Google Scholar] [CrossRef]
- Lee, J.; Vali, Y.; Boursier, J.; Spijker, R.; Anstee, Q.M.; Bossuyt, P.M.; Zafarmand, M.H. Prognostic accuracy of FIB-4, NAFLD fibrosis score and APRI for NAFLD-related events: A systematic review. Liver Int. Off. J. Int. Assoc. Study Liver 2021, 41, 261–270. [Google Scholar] [CrossRef]
- Yin, J.Y.; Yang, T.Y.; Yang, B.Q.; Hou, C.X.; Li, J.N.; Li, Y.; Wang, Q. FibroScan-aspartate transaminase: A superior non-invasive model for diagnosing high-risk metabolic dysfunction-associated steatohepatitis. World J. Gastroenterol. 2024, 30, 2440–2453. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yu, H.; Gao, J.; Yang, L.; Zhang, Y.; Yuan, X.; Zhang, Y. Machine learning deciphers the significance of mitochondrial regulators on the diagnosis and subtype classification in non-alcoholic fatty liver disease. Heliyon 2024, 10, e29860. [Google Scholar] [CrossRef] [PubMed]
- Cermelli, S.; Ruggieri, A.; Marrero, J.A.; Ioannou, G.N.; Beretta, L. Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS ONE 2011, 6, e23937. [Google Scholar] [CrossRef] [PubMed]
- Hendy, O.M.; Rabie, H.; El Fouly, A.; Abdel-Samiee, M.; Abdelmotelb, N.; Elshormilisy, A.A.; Allam, M.; Ali, S.T.; Bahaa El-Deen, N.M.; Abdelsattar, S.; et al. The Circulating Micro-RNAs (-122, -34a and -99a) as Predictive Biomarkers for Non-Alcoholic Fatty Liver Diseases. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 2715–2723. [Google Scholar] [CrossRef]
- Tan, Y.; Ge, G.; Pan, T.; Wen, D.; Gan, J. A pilot study of serum microRNAs panel as potential biomarkers for diagnosis of nonalcoholic fatty liver disease. PLoS ONE 2014, 9, e105192. [Google Scholar] [CrossRef]
- Liu, C.H.; Ampuero, J.; Gil-Gómez, A.; Montero-Vallejo, R.; Rojas, Á.; Muñoz-Hernández, R.; Gallego-Durán, R.; Romero-Gómez, M. miRNAs in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis. J. Hepatol. 2018, 69, 1335–1348. [Google Scholar] [CrossRef]
- Zhang, H.; Niu, Q.; Liang, K.; Li, X.; Jiang, J.; Bian, C. Effect of LncPVT1/miR-20a-5p on Lipid Metabolism and Insulin Resistance in NAFLD. Diabetes Metab. Syndr. Obes. Targets Ther. 2021, 14, 4599–4608. [Google Scholar] [CrossRef]
- Liu, X.L.; Pan, Q.; Zhang, R.N.; Shen, F.; Yan, S.Y.; Sun, C.; Xu, Z.J.; Chen, Y.W.; Fan, J.G. Disease-specific miR-34a as diagnostic marker of non-alcoholic steatohepatitis in a Chinese population. World J. Gastroenterol. 2016, 22, 9844–9852. [Google Scholar] [CrossRef]
- Xia, Y.; Zhang, Y.; Wang, H. Upregulated lncRNA HCG18 in Patients with Non-Alcoholic Fatty Liver Disease and Its Regulatory Effect on Insulin Resistance. Diabetes Metab. Syndr. Obes. Targets Ther. 2021, 14, 4747–4756. [Google Scholar] [CrossRef]
- Park, J.G.; Kim, G.; Jang, S.Y.; Lee, Y.R.; Lee, E.; Lee, H.W.; Han, M.H.; Chun, J.M.; Han, Y.S.; Yoon, J.S.; et al. Plasma Long Noncoding RNA LeXis is a Potential Diagnostic Marker for Non-Alcoholic Steatohepatitis. Life 2020, 10, 230. [Google Scholar] [CrossRef]
- Albadawy, R.; Agwa, S.H.A.; Khairy, E.; Saad, M.; El Touchy, N.; Othman, M.; Matboli, M. Clinical Significance of HSPD1/MMP14/ITGB1/miR-6881-5P/Lnc-SPARCL1-1:2 RNA Panel in NAFLD/NASH Diagnosis: Egyptian Pilot Study. Biomedicines 2021, 9, 1248. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Yuan, W.; Jiang, R.; Zhan, Z.; Zhang, L.; Xu, X.; Qian, Y.; Yang, W.; Zhang, Z. Machine learning-based integration identifies the ferroptosis hub genes in nonalcoholic steatohepatitis. Lipids Health Dis. 2024, 23, 23. [Google Scholar] [CrossRef] [PubMed]
- Deng, B.; Chen, Y.; He, P.; Liu, Y.; Li, Y.; Cai, Y.; Dong, W. Identification of Mitophagy-Associated Genes for the Prediction of Metabolic Dysfunction-Associated Steatohepatitis Based on Interpretable Machine Learning Models. J. Inflamm. Res. 2024, 17, 2711–2730. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Liu, W.; Guo, X.; Weng, S.; Chen, Y.; Luo, Y.; Wang, S.; Zhu, B.; Liu, Y.; Peng, D. Identification and validation of efferocytosis-related biomarkers for the diagnosis of metabolic dysfunction-associated steatohepatitis based on bioinformatics analysis and machine learning. Front. Immunol. 2024, 15, 1460431. [Google Scholar] [CrossRef]
- Ouyang, G.; Wu, Z.; Liu, Z.; Pan, G.; Wang, Y.; Liu, J.; Guo, J.; Liu, T.; Huang, G.; Zeng, Y.; et al. Identification and validation of potential diagnostic signature and immune cell infiltration for NAFLD based on cuproptosis-related genes by bioinformatics analysis and machine learning. Front. Immunol. 2023, 14, 1251750. [Google Scholar] [CrossRef]
- Qin, J.; Cao, P.; Ding, X.; Zeng, Z.; Deng, L.; Luo, L. Machine learning identifies ferroptosis-related gene ANXA2 as potential diagnostic biomarkers for NAFLD. Front. Endocrinol. 2023, 14, 1303426. [Google Scholar] [CrossRef]
- Fang, Z.; Liu, C.; Yu, X.; Yang, K.; Yu, T.; Ji, Y.; Liu, C. Identification of neutrophil extracellular trap-related biomarkers in non-alcoholic fatty liver disease through machine learning and single-cell analysis. Sci. Rep. 2024, 14, 21085. [Google Scholar] [CrossRef]
- Song, Y.M.; Ge, J.Y.; Ding, M.; Zheng, Y.W. Key factor screening in mouse NASH model using single-cell sequencing combined with machine learning. Heliyon 2024, 10, e33597. [Google Scholar] [CrossRef]
- Wan, Y.; Slevin, E.; Koyama, S.; Huang, C.K.; Shetty, A.K.; Li, X.; Harrison, K.; Li, T.; Zhou, B.; Lorenzo, S.R.; et al. miR-34a regulates macrophage-associated inflammation and angiogenesis in alcohol-induced liver injury. Hepatol. Commun. 2023, 7, e0089. [Google Scholar] [CrossRef]
- Rehati, A.; Abuduaini, B.; Liang, Z.; Chen, D.; He, F. Identification of heat shock protein family A member 5 (HSPA5) targets involved in nonalcoholic fatty liver disease. Genes. Immun. 2023, 24, 124–129. [Google Scholar] [CrossRef]
- Yokoyama, S.; Muto, H.; Honda, T.; Kurokawa, Y.; Ogawa, H.; Nakajima, R.; Kawashima, H.; Tani, H. Identification of Two Long Noncoding RNAs, Kcnq1ot1 and Rmst, as Biomarkers in Chronic Liver Diseases in Mice. Int. J. Mol. Sci. 2024, 25, 8927. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhao, M.; Miao, C.; Shi, X.; Lu, J. Identification and validation of key biomarkers associated with macrophages in nonalcoholic fatty liver disease based on hdWGCNA and machine learning. Aging 2023, 15, 15451–15472. [Google Scholar] [CrossRef] [PubMed]
- Rozanova, S.; Barkovits, K.; Nikolov, M.; Schmidt, C.; Urlaub, H.; Marcus, K. Quantitative Mass Spectrometry-Based Proteomics: An Overview. Methods Mol. Biol. 2021, 2228, 85–116. [Google Scholar] [CrossRef] [PubMed]
- Stiglund, N.; Hagström, H.; Stål, P.; Cornillet, M.; Björkström, N.K. Dysregulated peripheral proteome reveals NASH-specific signatures identifying patient subgroups with distinct liver biology. Front. Immunol. 2023, 14, 1186097. [Google Scholar] [CrossRef]
- Jia, X.; Song, E.; Liu, Y.; Chen, J.; Wan, P.; Hu, Y.; Ye, D.; Chakrabarti, S.; Mahajan, H.; George, J.; et al. Identification and multicentric validation of soluble CDCP1 as a robust serological biomarker for risk stratification of NASH in obese Chinese. Cell Reports. Med. 2023, 4, 101257. [Google Scholar] [CrossRef]
- Ding, J.; Liu, H.; Zhang, X.; Zhao, N.; Peng, Y.; Shi, J.; Chen, J.; Chi, X.; Li, L.; Zhang, M.; et al. Integrative multiomic analysis identifies distinct molecular subtypes of NAFLD in a Chinese population. Sci. Transl. Med. 2024, 16, eadh9940. [Google Scholar] [CrossRef]
- Aregay, A.; Engel, B.; Port, K.; Vondran, F.W.R.; Bremer, B.; Niehaus, C.; Khera, T.; Richter, N.; Jaeckel, E.; Cornberg, M.; et al. Distinct Immune Imprints of Post-Liver Transplantation Hepatitis C Persist Despite Viral Clearance. Liver Transplant. Off. Publ. Am. Assoc. Study Liver Dis. Int. Liver Transplant. Soc. 2021, 27, 887–899. [Google Scholar] [CrossRef]
- Molaei, V.; Fattahi, M.R.; Haghshenas, M.R.; Hosseini, S.Y.; Malekhosseini, S.A.; Sarvari, J. Polymorphism Analysis of Interleukin-18 and Interleukin-37 Genes in Hepatitis B Infections with Different Outcomes: A Preliminary Report from an Iranian Population. Asian Pac. J. Cancer Prev. APJCP 2023, 24, 411–416. [Google Scholar] [CrossRef]
- Taru, V.; Szabo, G.; Mehal, W.; Reiberger, T. Inflammasomes in chronic liver disease: Hepatic injury, fibrosis progression and systemic inflammation. J. Hepatol. 2024, 81, 895–910. [Google Scholar] [CrossRef]
- Veyel, D.; Wenger, K.; Broermann, A.; Bretschneider, T.; Luippold, A.H.; Krawczyk, B.; Rist, W.; Simon, E. Biomarker discovery for chronic liver diseases by multi-omics—A preclinical case study. Sci. Rep. 2020, 10, 1314. [Google Scholar] [CrossRef]
- Govaere, O.; Hasoon, M.; Alexander, L.; Cockell, S.; Tiniakos, D.; Ekstedt, M.; Schattenberg, J.M.; Boursier, J.; Bugianesi, E.; Ratziu, V.; et al. A proteo-transcriptomic map of non-alcoholic fatty liver disease signatures. Nat. Metab. 2023, 5, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Sveinbjornsson, G.; Ulfarsson, M.O.; Thorolfsdottir, R.B.; Jonsson, B.A.; Einarsson, E.; Gunnlaugsson, G.; Rognvaldsson, S.; Arnar, D.O.; Baldvinsson, M.; Bjarnason, R.G.; et al. Multiomics study of nonalcoholic fatty liver disease. Nat. Genet. 2022, 54, 1652–1663. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Wadhawan, S.; Greenfield, A.; Decato, B.E.; Oseini, A.M.; Collen, R.; Shevell, D.E.; Thompson, J.; Jarai, G.; Charles, E.D.; et al. SOMAscan Proteomics Identifies Serum Biomarkers Associated With Liver Fibrosis in Patients With NASH. Hepatol. Commun. 2021, 5, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Adamek, A.; Kasprzak, A. Insulin-Like Growth Factor (IGF) System in Liver Diseases. Int. J. Mol. Sci. 2018, 19, 1308. [Google Scholar] [CrossRef]
- Niu, L.; Geyer, P.E.; Wewer Albrechtsen, N.J.; Gluud, L.L.; Santos, A.; Doll, S.; Treit, P.V.; Holst, J.J.; Knop, F.K.; Vilsbøll, T.; et al. Plasma proteome profiling discovers novel proteins associated with non-alcoholic fatty liver disease. Mol. Syst. Biol. 2019, 15, e8793. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, X.; Xu, M.; Yue, T.; Ling, P.; Fang, T.; Luo, S.; Xu, S.; Weng, J. Comparative Proteomic Analysis of Liver Tissues and Serum in db/db Mice. Int. J. Mol. Sci. 2022, 23, 9687. [Google Scholar] [CrossRef]
- Liu, T.; Liu, D.; Liu, R.; Jiang, H.; Yan, G.; Li, W.; Sun, L.; Zhang, S.; Liu, Y.; Guo, K. Discovering potential serological biomarker for chronic Hepatitis B Virus-related hepatocellular carcinoma in Chinese population by MAL-associated serum glycoproteomics analysis. Sci. Rep. 2017, 7, 38918. [Google Scholar] [CrossRef]
- Pyo, J.J.; Choi, Y. Key hepatic signatures of human and mouse nonalcoholic steatohepatitis: A transcriptome-proteome data meta-analysis. Front. Endocrinol. 2022, 13, 934847. [Google Scholar] [CrossRef]
- da Silva Lima, N.; Fondevila, M.F.; Nóvoa, E.; Buqué, X.; Mercado-Gómez, M.; Gallet, S.; González-Rellan, M.J.; Fernandez, U.; Loyens, A.; Garcia-Vence, M.; et al. Inhibition of ATG3 ameliorates liver steatosis by increasing mitochondrial function. J. Hepatol. 2022, 76, 11–24. [Google Scholar] [CrossRef]
- Khoury, M.; Guo, Q.; Furuta, K.; Correia, C.; Meroueh, C.; Kim Lee, H.S.; Warasnhe, K.; Valenzuela-Pérez, L.; Mazar, A.P.; Kim, I.; et al. Glycogen synthase kinase 3 activity enhances liver inflammation in MASH. JHEP Rep. Innov. Hepatol. 2024, 6, 101073. [Google Scholar] [CrossRef]
- Aslam, B.; Basit, M.; Nisar, M.A.; Khurshid, M.; Rasool, M.H. Proteomics: Technologies and Their Applications. J. Chromatogr. Sci. 2017, 55, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Angelini, G.; Panunzi, S.; Castagneto-Gissey, L.; Pellicanò, F.; De Gaetano, A.; Pompili, M.; Riccardi, L.; Garcovich, M.; Raffaelli, M.; Ciccoritti, L.; et al. Accurate liquid biopsy for the diagnosis of non-alcoholic steatohepatitis and liver fibrosis. Gut 2023, 72, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Indira Chandran, V.; Wernberg, C.W.; Lauridsen, M.M.; Skytthe, M.K.; Bendixen, S.M.; Larsen, F.T.; Hansen, C.D.; Grønkjær, L.L.; Siersbæk, M.S.; Caterino, T.D.; et al. Circulating TREM2 as a noninvasive diagnostic biomarker for NASH in patients with elevated liver stiffness. Hepatology 2023, 77, 558–572. [Google Scholar] [CrossRef] [PubMed]
- Kothari, V.; Savard, C.; Tang, J.; Lee, S.P.; Subramanian, S.; Wang, S.; den Hartigh, L.J.; Bornfeldt, K.E.; Ioannou, G.N. sTREM2 is a plasma biomarker for human NASH and promotes hepatocyte lipid accumulation. Hepatol. Commun. 2023, 7, e0265. [Google Scholar] [CrossRef]
- Wu, X.; Cheung, C.K.Y.; Ye, D.; Chakrabarti, S.; Mahajan, H.; Yan, S.; Song, E.; Yang, W.; Lee, C.H.; Lam, K.S.L.; et al. Serum Thrombospondin-2 Levels Are Closely Associated With the Severity of Metabolic Syndrome and Metabolic Associated Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2022, 107, e3230–e3240. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Williams, S.A.; Lavine, J.E.; Neuschwander-Tetri, B.A.; Alexander, L.; Ostroff, R.; Biegel, H.; Kowdley, K.V.; Chalasani, N.; Dasarathy, S.; et al. Defining the serum proteomic signature of hepatic steatosis, inflammation, ballooning and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2023, 78, 693–703. [Google Scholar] [CrossRef]
- Corey, K.E.; Pitts, R.; Lai, M.; Loureiro, J.; Masia, R.; Osganian, S.A.; Gustafson, J.L.; Hutter, M.M.; Gee, D.W.; Meireles, O.R.; et al. ADAMTSL2 protein and a soluble biomarker signature identify at-risk non-alcoholic steatohepatitis and fibrosis in adults with NAFLD. J. Hepatol. 2022, 76, 25–33. [Google Scholar] [CrossRef]
- Feng, G.; Zhang, X.; Zhang, L.; Liu, W.Y.; Geng, S.; Yuan, H.Y.; Sha, J.C.; Wang, X.D.; Sun, D.Q.; Targher, G.; et al. Novel urinary protein panels for the non-invasive diagnosis of non-alcoholic fatty liver disease and fibrosis stages. Liver Int. Off. J. Int. Assoc. Study Liver 2023, 43, 1234–1246. [Google Scholar] [CrossRef]
- Sourianarayanane, A.; Salemi, M.R.; Phinney, B.S.; McCullough, A.J. Liver Tissue Proteins Improve the Accuracy of Plasma Proteins as Biomarkers in Diagnosing Metabolic Dysfunction-Associated Steatohepatitis. Proteomics. Clin. Appl. 2024, 18, e202300236. [Google Scholar] [CrossRef]
- Sydor, S.; Dandyk, C.; Schwerdt, J.; Manka, P.; Benndorf, D.; Lehmann, T.; Schallert, K.; Wolf, M.; Reichl, U.; Canbay, A.; et al. Discovering Biomarkers for Non-Alcoholic Steatohepatitis Patients with and without Hepatocellular Carcinoma Using Fecal Metaproteomics. Int. J. Mol. Sci. 2022, 23, 8841. [Google Scholar] [CrossRef]
- Newgard, C.B. Metabolomics and Metabolic Diseases: Where Do We Stand? Cell Metab. 2017, 25, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Gross, R.W. The foundations and development of lipidomics. J. Lipid Res. 2022, 63, 100164. [Google Scholar] [CrossRef] [PubMed]
- Rappez, L.; Stadler, M.; Triana, S.; Gathungu, R.M.; Ovchinnikova, K.; Phapale, P.; Heikenwalder, M.; Alexandrov, T. SpaceM reveals metabolic states of single cells. Nat. Methods 2021, 18, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Ščupáková, K.; Soons, Z.; Ertaylan, G.; Pierzchalski, K.A.; Eijkel, G.B.; Ellis, S.R.; Greve, J.W.; Driessen, A.; Verheij, J.; De Kok, T.M.; et al. Spatial Systems Lipidomics Reveals Nonalcoholic Fatty Liver Disease Heterogeneity. Anal. Chem. 2018, 90, 5130–5138. [Google Scholar] [CrossRef]
- Stopka, S.A.; van der Reest, J.; Abdelmoula, W.M.; Ruiz, D.F.; Joshi, S.; Ringel, A.E.; Haigis, M.C.; Agar, N.Y.R. Spatially resolved characterization of tissue metabolic compartments in fasted and high-fat diet livers. PLoS ONE 2022, 17, e0261803. [Google Scholar] [CrossRef]
- Montefusco, D.; Lambert, J.; Anderson, A.; Allegood, J.; Cowart, L.A. Analysis of the Sphingolipidome in NAFLD. Methods Mol. Biol. 2022, 2455, 279–303. [Google Scholar] [CrossRef]
- Yadav, A.K.; MacNeill, J.J.; Krylov, A.; Ashrafi, N.; Mimi, R.A.; Saxena, R.; Liu, S.; Graham, S.F.; Wan, J.; Morral, N. Sex- and age-associated factors drive the pathophysiology of MASLD. Hepatol. Commun. 2024, 8, e0523. [Google Scholar] [CrossRef]
- Ooi, G.J.; Meikle, P.J.; Huynh, K.; Earnest, A.; Roberts, S.K.; Kemp, W.; Parker, B.L.; Brown, W.; Burton, P.; Watt, M.J. Hepatic lipidomic remodeling in severe obesity manifests with steatosis and does not evolve with non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 524–535. [Google Scholar] [CrossRef]
- Montandon, S.A.; Somm, E.; Loizides-Mangold, U.; de Vito, C.; Dibner, C.; Jornayvaz, F.R. Multi-technique comparison of atherogenic and MCD NASH models highlights changes in sphingolipid metabolism. Sci. Rep. 2019, 9, 16810. [Google Scholar] [CrossRef]
- Mann, J.P.; Jenkins, B.; Furse, S.; Snowden, S.G.; Alisi, A.; Draijer, L.G.; Karnebeek, K.; Kelly, D.A.; Koot, B.G.; Mosca, A.; et al. Comparison of the Lipidomic Signature of Fatty Liver in Children and Adults: A Cross-Sectional Study. J. Pediatr. Gastroenterol. Nutr. 2022, 74, 734–741. [Google Scholar] [CrossRef]
- Yang, J.; Dai, M.; Wang, Y.; Yan, Z.; Mao, S.; Liu, A.; Lu, C. A CDAHFD-induced mouse model mimicking human NASH in the metabolism of hepatic phosphatidylcholines and acyl carnitines. Food Funct. 2024, 15, 2982–2995. [Google Scholar] [CrossRef] [PubMed]
- Fitzinger, J.; Rodriguez-Blanco, G.; Herrmann, M.; Borenich, A.; Stauber, R.; Aigner, E.; Mangge, H. Gender-Specific Bile Acid Profiles in Non-Alcoholic Fatty Liver Disease. Nutrients 2024, 16, 250. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Muthiah, M.D.; Narayan, N.; Siddiqui, M.S.; Puri, P.; Luketic, V.A.; Contos, M.J.; Idowu, M.; Chuang, J.C.; Billin, A.N.; et al. Metabolic reprogramming of the intestinal microbiome with functional bile acid changes underlie the development of NAFLD. Hepatology 2022, 76, 1811–1824. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhao, L.; Xiong, X.; He, Y.; Huang, W.; Liu, Z.; Ji, L.; Pan, B.; Guo, X.; Wang, L.; et al. TMAVA, a Metabolite of Intestinal Microbes, Is Increased in Plasma From Patients With Liver Steatosis, Inhibits γ-Butyrobetaine Hydroxylase, and Exacerbates Fatty Liver in Mice. Gastroenterology 2020, 158, 2266–2281.e27. [Google Scholar] [CrossRef]
- Fuchs, C.D.; Simbrunner, B.; Baumgartner, M.; Campbell, C.; Reiberger, T.; Trauner, M. Bile acid metabolism and signalling in liver disease. J. Hepatol. 2025, 82, 134–153. [Google Scholar] [CrossRef]
- Ganesan, R.; Gupta, H.; Jeong, J.J.; Sharma, S.P.; Won, S.M.; Oh, K.K.; Yoon, S.J.; Kim, D.J.; Suk, K.T. A metabolomics approach to the validation of predictive metabolites and phenotypic expression in non-alcoholic fatty liver disease. Life Sci. 2023, 322, 121626. [Google Scholar] [CrossRef]
- McGlinchey, A.J.; Govaere, O.; Geng, D.; Ratziu, V.; Allison, M.; Bousier, J.; Petta, S.; de Oliviera, C.; Bugianesi, E.; Schattenberg, J.M.; et al. Metabolic signatures across the full spectrum of non-alcoholic fatty liver disease. JHEP Rep. Innov. Hepatol. 2022, 4, 100477. [Google Scholar] [CrossRef]
- Gautam, J.; Aggarwal, H.; Kumari, D.; Gupta, S.K.; Kumar, Y.; Dikshit, M. A methionine-choline-deficient diet induces nonalcoholic steatohepatitis and alters the lipidome, metabolome, and gut microbiome profile in the C57BL/6J mouse. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2024, 1869, 159545. [Google Scholar] [CrossRef]
- Fotakis, C.; Kalafati, I.P.; Amanatidou, A.I.; Andreou, V.; Matzapetakis, M.; Kafyra, M.; Varlamis, I.; Zervou, M.; Dedoussis, G.V. Serum metabolomic profiling unveils distinct sex-related metabolic patterns in NAFLD. Front. Endocrinol. 2023, 14, 1230457. [Google Scholar] [CrossRef]
- Yan, Y.; Chen, Q.; Xiang, Z.; Wang, Q.; Long, Z.; Liang, H.; Ameer, S.; Zou, J.; Dai, X.; Zhu, Z. Amino acid metabolomics and machine learning-driven assessment of future liver remnant growth after hepatectomy in livers of various backgrounds. J. Pharm. Biomed. Anal. 2024, 249, 116369. [Google Scholar] [CrossRef]
- Lanekoff, I.; Thomas, M.; Laskin, J. Shotgun approach for quantitative imaging of phospholipids using nanospray desorption electrospray ionization mass spectrometry. Anal. Chem. 2014, 86, 1872–1880. [Google Scholar] [CrossRef] [PubMed]
- Noureddin, M.; Truong, E.; Mayo, R.; Martínez-Arranz, I.; Mincholé, I.; Banales, J.M.; Arrese, M.; Cusi, K.; Arias-Loste, M.T.; Bruha, R.; et al. Serum identification of at-risk MASH: The metabolomics-advanced steatohepatitis fibrosis score (MASEF). Hepatology 2024, 79, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Iruzubieta, P.; Mayo, R.; Mincholé, I.; Martínez-Arranz, I.; Arias-Loste, M.T.; Ibañez-Samaniego, L.; Ampuero, J.; Abad, J.; Martín-Mateos, R.; Fernández-Laso, A.B.; et al. One-step non-invasive diagnosis of metabolic dysfunction-associated steatohepatitis and fibrosis in high-risk population. United Eur. Gastroenterol. J. 2024, 12, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Quehenberger, O.; Armando, A.M.; Cedeno, T.H.; Loomba, R.; Sanyal, A.J.; Dennis, E.A. Novel eicosanoid signature in plasma provides diagnostic for metabolic dysfunction-associated steatotic liver disease. J. Lipid Res. 2024, 65, 100647. [Google Scholar] [CrossRef]
- Jung, Y.; Lee, M.K.; Puri, P.; Koo, B.K.; Joo, S.K.; Jang, S.Y.; Lee, D.H.; Jung, Y.J.; Kim, B.G.; Lee, K.L.; et al. Circulating lipidomic alterations in obese and non-obese subjects with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2020, 52, 1603–1614. [Google Scholar] [CrossRef]
- Zhu, Q.; Li, H.; Ao, Z.; Xu, H.; Luo, J.; Kaurich, C.; Yang, R.; Zhu, P.W.; Chen, S.D.; Wang, X.D.; et al. Lipidomic identification of urinary extracellular vesicles for non-alcoholic steatohepatitis diagnosis. J. Nanobiotechnol. 2022, 20, 349. [Google Scholar] [CrossRef]
- Mouskeftara, T.; Kalopitas, G.; Liapikos, T.; Arvanitakis, K.; Germanidis, G.; Gika, H. Predicting Non-Alcoholic Steatohepatitis: A Lipidomics-Driven Machine Learning Approach. Int. J. Mol. Sci. 2024, 25, 5965. [Google Scholar] [CrossRef]
- Zhou, Y.; Orešič, M.; Leivonen, M.; Gopalacharyulu, P.; Hyysalo, J.; Arola, J.; Verrijken, A.; Francque, S.; Van Gaal, L.; Hyötyläinen, T.; et al. Noninvasive Detection of Nonalcoholic Steatohepatitis Using Clinical Markers and Circulating Levels of Lipids and Metabolites. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2016, 14, 1463–1472.e6. [Google Scholar] [CrossRef]
- Carr, R.M.; Li, Y.; Chau, L.; Friedman, E.S.; Lee, J.J.; Adorini, L.; Erickson, M.; Zaru, L.; Shringarpure, R.; MacConell, L.; et al. An integrated analysis of fecal microbiome and metabolomic features distinguish non-cirrhotic NASH from healthy control populations. Hepatology 2023, 78, 1843–1857. [Google Scholar] [CrossRef]
- Ji, M.; Jo, Y.; Choi, S.J.; Kim, S.M.; Kim, K.K.; Oh, B.C.; Ryu, D.; Paik, M.J.; Lee, D.H. Plasma Metabolomics and Machine Learning-Driven Novel Diagnostic Signature for Non-Alcoholic Steatohepatitis. Biomedicines 2022, 10, 1669. [Google Scholar] [CrossRef]
- Leung, H.; Long, X.; Ni, Y.; Qian, L.; Nychas, E.; Siliceo, S.L.; Pohl, D.; Hanhineva, K.; Liu, Y.; Xu, A.; et al. Risk assessment with gut microbiome and metabolite markers in NAFLD development. Sci. Transl. Med. 2022, 14, eabk0855. [Google Scholar] [CrossRef] [PubMed]
- Vvedenskaya, O.; Rose, T.D.; Knittelfelder, O.; Palladini, A.; Wodke, J.A.H.; Schuhmann, K.; Ackerman, J.M.; Wang, Y.; Has, C.; Brosch, M.; et al. Nonalcoholic fatty liver disease stratification by liver lipidomics. J. Lipid Res. 2021, 62, 100104. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Zheng, K.I.; Wang, X.D.; Qiao, J.; Li, Y.Y.; Zhang, L.; Zheng, M.H.; Wu, J. LC-MS-based lipidomic analysis in distinguishing patients with nonalcoholic steatohepatitis from nonalcoholic fatty liver. Hepatobiliary Pancreat. Dis. Int. HBPD INT 2021, 20, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Beger, R.D.; Dunn, W.; Schmidt, M.A.; Gross, S.S.; Kirwan, J.A.; Cascante, M.; Brennan, L.; Wishart, D.S.; Oresic, M.; Hankemeier, T.; et al. Metabolomics enables precision medicine: “A White Paper, Community Perspective”. Metabolomics Off. J. Metabolomic Soc. 2016, 12, 149. [Google Scholar] [CrossRef]
- Szabo, Z.; Janaky, T. Challenges and developments in protein identification using mass spectrometry. TrAC Trends Anal. Chem. 2015, 69, 76–87. [Google Scholar] [CrossRef]
- Thiele, M.; Villesen, I.F.; Niu, L.; Johansen, S.; Sulek, K.; Nishijima, S.; Espen, L.V.; Keller, M.; Israelsen, M.; Suvitaival, T.; et al. Opportunities and barriers in omics-based biomarker discovery for steatotic liver diseases. J. Hepatol. 2024, 81, 345–359. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| RNA Transcript | Comparisons | Prediction Models | Sensitivity | Specificity | AUROC | Refs. |
|---|---|---|---|---|---|---|
| miRNA |
| MiR-122 | NA | NA |
| [83] |
| NAFLD—simple steatosis vs. NASH | MiR-122 | 92% | 85% | 0.81 | [84] | |
| NAFLD—simple steatosis vs. NASH | MiR-122 | NA | NA | 0.71 | [58] | |
| NAFLD vs. C | MiR-122-5p, miR-1290, miR-27b-3p, and miR-192-5p | 69.9% | 83.7% | 0.78 | [85] | |
| NAFLD vs. C | MiR-122 | NA | NA | 0.82 | [86] | |
| NAFLD vs. C | MiR-20a-5p | 84% | 84.6% | 0.89 | [87] | |
| NASH vs. C | MiR-34a | 70.4% | 68.7% | 0.81 | [88] | |
| lncRNA | NASH with F3-4 vs. NASH with F0-2 | TGFB2/TGFB2-OT1, and FIB-4 TGFB2/TGFB2-OT1, and FibroScan | NA | NA | 0.891 0.892 | [76] |
| NAFLD vs. C | LncRNA HCG18 and miR-197-3p | NA | NA | 0.93 | [89] | |
| NAFLD vs. NASH | LncRNA LeXis | 54.3% | 100% | 0.74 | [90] | |
| LncRNA HSPD1 lncRNA MMP14 lncRNA ITGB1 lncRNA SPARCL1-1:2 miR-6881-5p |
|
|
| [91] | |
| mRNA | NAFLD vs. C | AKR1B10, TYMS, and TREM2 | NA | NA | 0.71 | [92] |
| MASH vs. C | MRAS, RAB7B, and RETREG1 | NA | NA | 0.93 | [93] | |
| NAFLD vs. C | BCL2L11 NAGS RMND HDHD3 | NA | NA | 0.95 0.93 0.98 0.96 | [82] | |
| NAFLD vs. C | TREM2 TIMD4 | NA | NA | 0.95 0.96 | [94] | |
| NAFLD vs. C | NDUFA4 TFAM CDKN1B | NA | NA | 0.93 0.90 0.91 | [82] | |
| NAFLD vs. C | NFE2L2, DLD, and POLD1 | NA | NA | 0.70 | [95] | |
| NAFLD vs. C | ANXA2 | NA | NA | 0.95 | [96] | |
| NASH vs. C | PHLDA1 ZFP36L2 | NA | NA | 0.78 0.71 | [97] | |
| NAFLD vs. severe NAFLD | IL32-ALT-AST | NA | NA | 0.92 | [30] | |
| NASH vs. C | Anax1 and Gpnmb | 87.5% | 59.6% | 0.81 | [98] |
| Function | Comparisons | Prediction Models | Sensitivity | Specificity | AUROC | Refs. |
|---|---|---|---|---|---|---|
| lipid metabolism and cell signaling | NASH vs. C | PLIN2-Diabetes-Triglycerides-ALT-waist circumference RAB14 | 88% 96.9% | 100% 34.5% | 0.976 0.824 | [122] |
| immunomodulation | simple steatosis vs. MASH | sTREM2 | NA | NA | 0.8 | [111] |
| NASH vs. C | sTREM2 | 54% | 89% | 0.83 | [123] | |
| NAFL vs. NASH | sTREM2 | NA | NA | 0.80 | [124] | |
| extracellular matrix and cell–cell adhesion | benign steatosis vs. MASH | TSP2 | NA | NA | 0.84 | [125] |
| metabolic enzyme | NASH vs. C | PTGR1 | 87% | 63% | 0.85 | [126] |
| extracellular matrix formation and cell signaling |
| ADAMTSL2 |
|
|
| [127] |
| cell signaling, extracellular matrix formation, and cell signaling |
|
| NA | NA |
| [128] |
| cell growth, differentiation, and fibrosis | MASH vs. C | Fibrinogen α, IGFBP-3, COX4-1, HBP1, IGF-1R, VEGFR-2, EGFR, PDGFR-β, and Fibrinogen β | NA | NA | 0.79 | [129] |
| Metabolite | Comparisons | Prediction Models | Sensitivity | Specificity | AUROC | Refs. |
| lipid and fatty acid | High-risk MASH vs. no-risk MASH | MASEF (2 triglycerides, 5 glycerophosphocholines, 1 cholesteryl ester, 1 ceramide, and 3 sphingomyelins) | 78% | 65% | 0.79 | [152] |
| simple steatosis vs. MASH | OWLiver Panel (OWLiver DM2 and MASEF) | 86% | 35% | 0.788 | [153] | |
| MASLD vs. MASH | DAG 32:1, DAG 34:0, DAG 34:1, TAG 40:1, TAG 44:1, TAG.48:1, TAG 50:2, and SM d36:0 | NA | NA | 0.808 | [155] | |
| MASLD vs. C | 4 HDoHE, 14 HDoHE, 5-HETE, 12-HETE, 15-HETE, 12-HEPE, 5,6-EET, 11,12-EET, 14,15-EET, 15-HETrE, 9,10-diHOME, 9-HODE, DHA, EPA, and adrenic acid | NA | NA | 0.999 | [154] | |
| NAFL vs. NASH | FFA (18:0), LPC (22:6/0:0), FFA (18:1), and PI (16:0/18:1) | NA | NA | 0.923 | [156] | |
| HOMA-IR, BMI, platelets count, LDL-c, ferritin, AST, FA 12:0, FA 18:3 ω3, FA 20:4 ω6/FA 20:5 ω3, CAR 4:0, LPC 20:4, LPC O-16:1, LPE 18:0, DG 18:1_18:2, and CE 20:4 |
|
|
| [157] | |
| amino acid and lipid | MASLD vs. MASH | glutamate, isoleucine, glycine, lysophosphatidylcholine 16:0, phosphoethanolamine 40:6, AST, and fasting insulin, along with PNPLA3 genotype | 86% | 35% | 0.866 | [158] |
| bile acid | MASH vs. C | Stool GDCA, Stool DCA, Stool GCDCA, Stool TLCA, Stool CDCA, Stool CA, Stool TDCA, Stool LCA, Stool GCA, Stool TCA, Stool TCDCA, Stool GLCA, age, and BMI | NA | NA | 0.986 | [159] |
| amino acid |
| MetaNASH (glutamic acid, isocitric acid, and aspartic acid) | NA | NA |
| [160] |
| organic acid | Mild steatosis vs. severe steatosis | Phenyllactic acid, hydrocinnamic acid, methanobrevibacter, fasting insulin, L-valine, age, 8,11,14-eicosatrienoic acid, suberic acid, BMI, 2-phenylpropionate, HDL, N-acetylserotonin, oxoglutaric acid, N-acetyltryptophan, PWY-3801, PWY-6167, PWY-7345, and Slackia | NA | NA | 0.78 | [161] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Bian, S.; Le, J. Molecular Landscape and Diagnostic Model of MASH: Transcriptomic, Proteomic, Metabolomic, and Lipidomic Perspectives. Genes 2025, 16, 399. https://doi.org/10.3390/genes16040399
Chen Y, Bian S, Le J. Molecular Landscape and Diagnostic Model of MASH: Transcriptomic, Proteomic, Metabolomic, and Lipidomic Perspectives. Genes. 2025; 16(4):399. https://doi.org/10.3390/genes16040399
Chicago/Turabian StyleChen, Yilong, Shuixiu Bian, and Jiamei Le. 2025. "Molecular Landscape and Diagnostic Model of MASH: Transcriptomic, Proteomic, Metabolomic, and Lipidomic Perspectives" Genes 16, no. 4: 399. https://doi.org/10.3390/genes16040399
APA StyleChen, Y., Bian, S., & Le, J. (2025). Molecular Landscape and Diagnostic Model of MASH: Transcriptomic, Proteomic, Metabolomic, and Lipidomic Perspectives. Genes, 16(4), 399. https://doi.org/10.3390/genes16040399