
Clinical Significance of Early-Onset Alzheimer’s Mutations in Asian and Western Populations: A Scoping Review

Abstract
1. Introduction
2. Materials and Methods
2.1. Search Strategy
2.2. Study Selection
2.3. Sampling Design
2.3.1. Inclusion Criteria
2.3.2. Exclusion Criteria
- (a)
- Types of articles other than original research papers, such as abstracts, review papers, conference proceedings, book or book chapters;
- (b)
- Articles that were not in the English language;
- (c)
- Articles published before January 2016; we opted to review articles published from 2016 onwards because the MeSH terms used in our search showed a higher number of publications beginning in that year, coinciding with the introduction of next-generation sequencing technologies. Additionally, the primary focus of this review was to compare genetic findings in EOAD between Asian and Western cohorts;
- (d)
- Studies on mutations other than APP, PSEN1, or PSEN;
- (e)
- Alzheimer’s research solely focused on late-onset AD, as this would not reflect EOAD;
- (f)
- Articles concerning dementia other than those related to Alzheimer’s (other forms/causes of dementia, i.e., Lewy body, vascular, frontotemporal, Parkinson’s, Huntington’s).
2.4. Identifying Research Questions and Relevant Studies
2.5. Assessing Studies for Eligibility
2.6. Data Analysis
3. Results
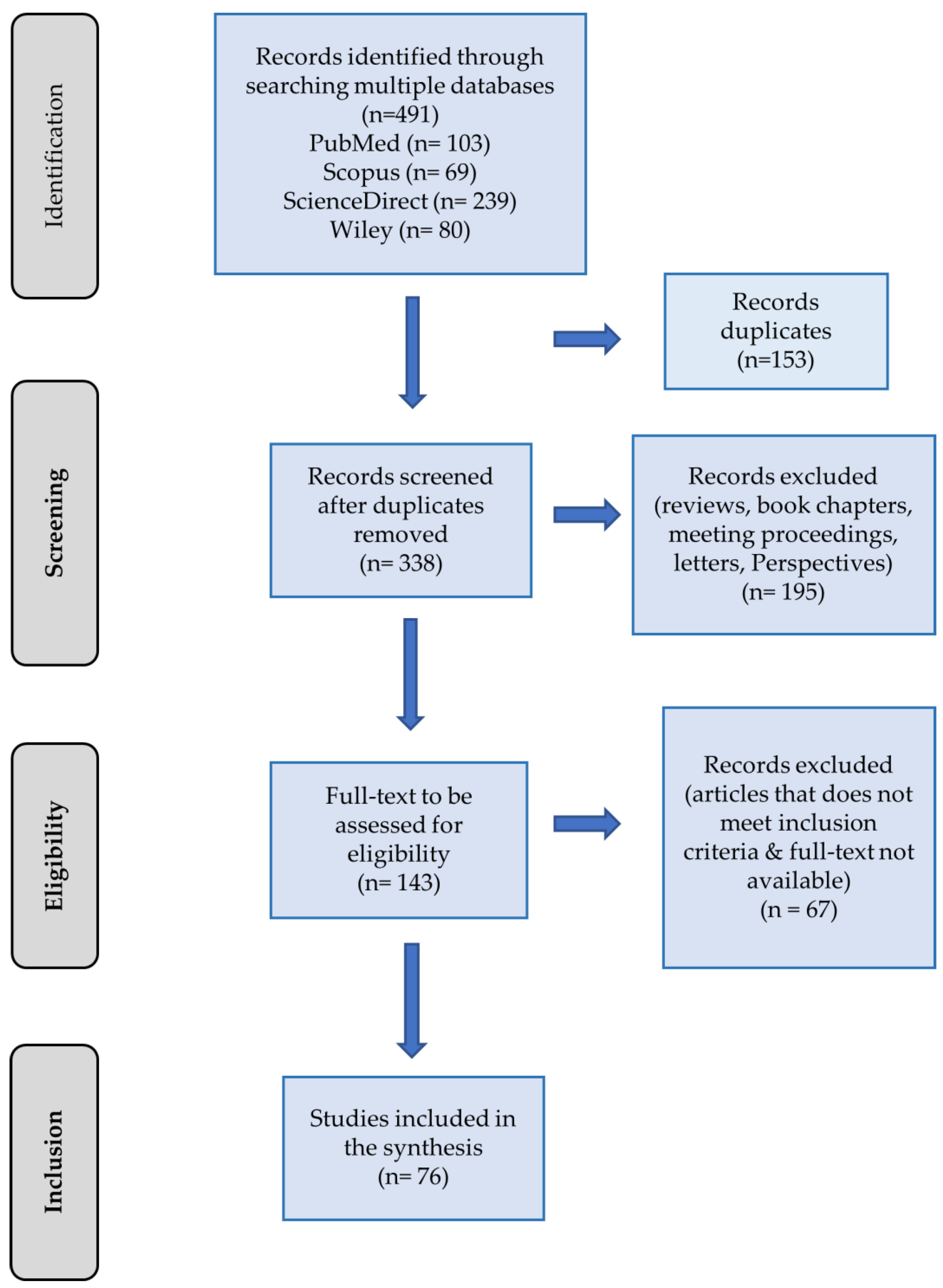
3.1. Selection of Studies
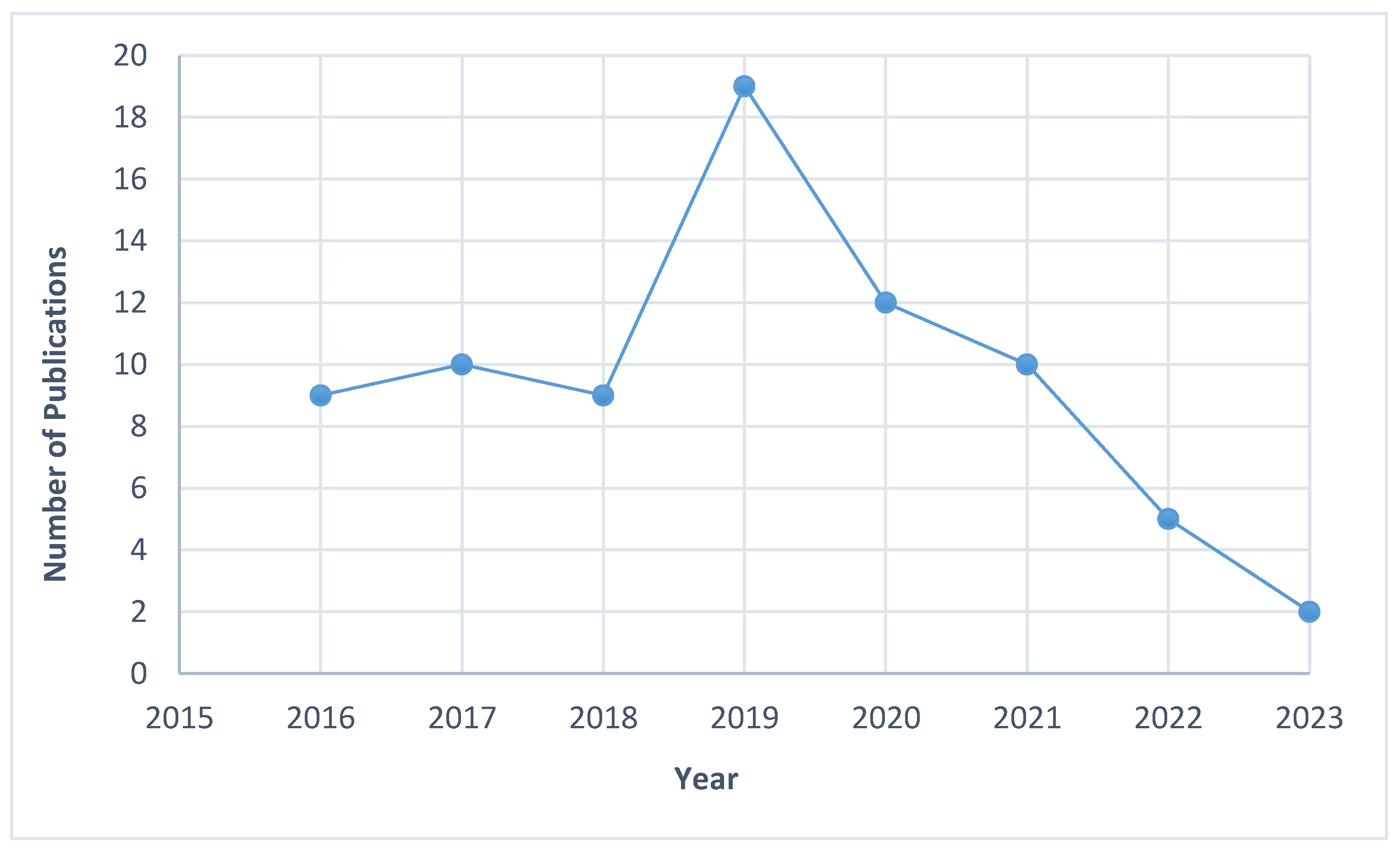
3.2. Characteristics of the Studies
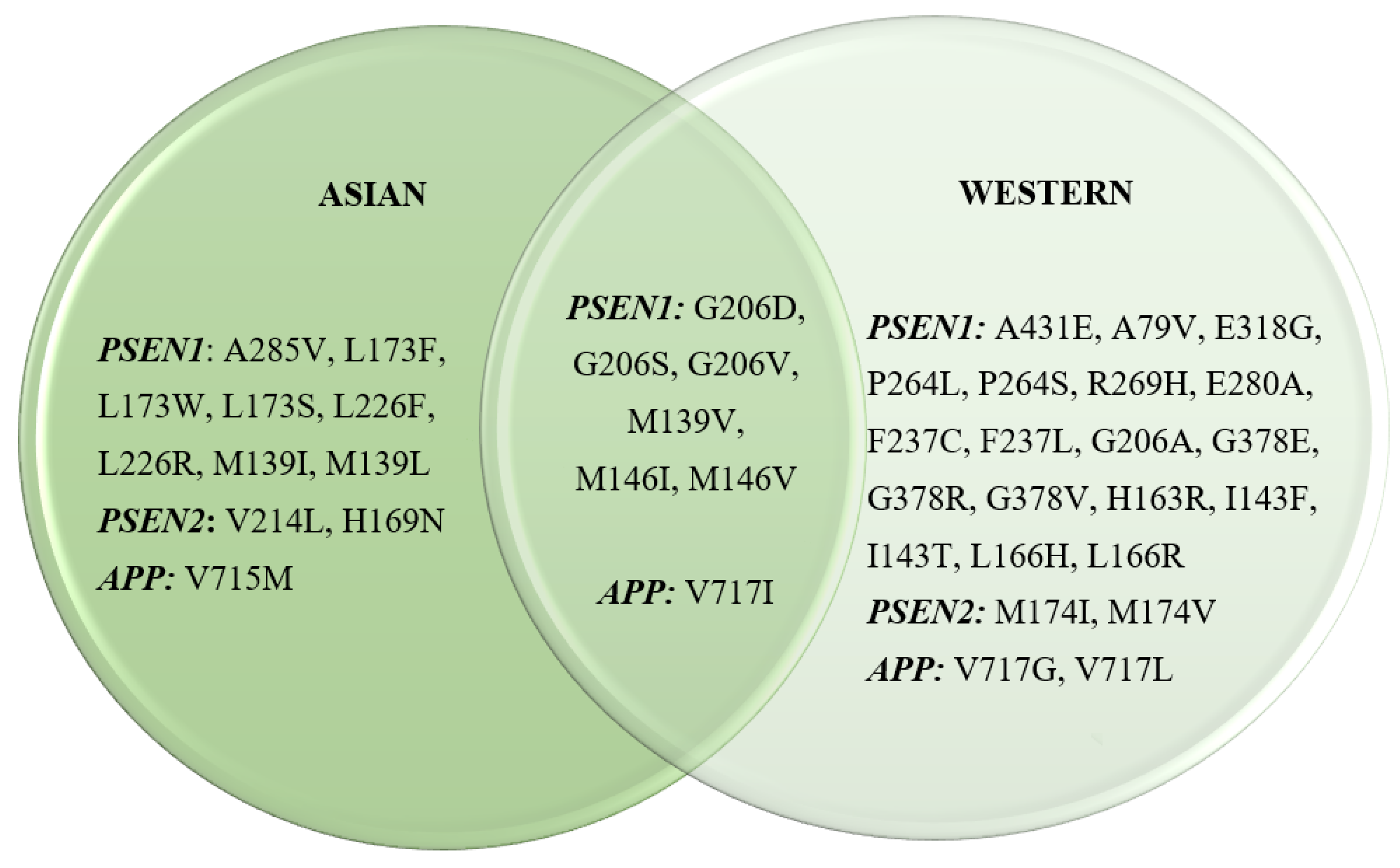
3.3. Common Mutations Identified in Asian and Western Populations
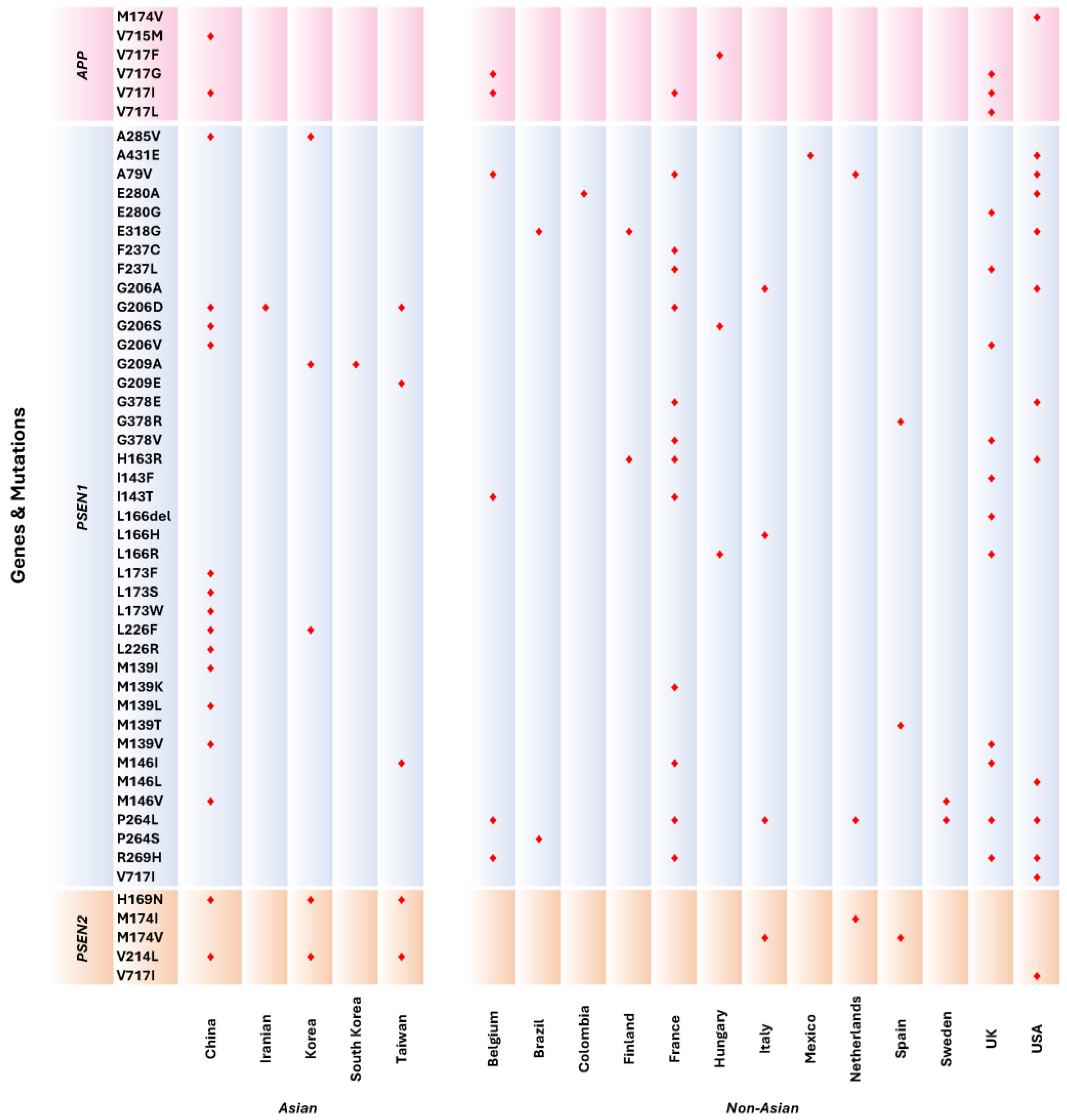
3.4. Distribution of APP, PSEN1, and PSEN2 Mutations in Asian and Western Populations
3.5. Clinical Phenotypes for Shared Mutations Observed in Both Asian and Western Populations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deture, M.A.; Dickson, D.W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H. Dementia Epidemiology Fact Sheet 2022. Ann. Rehabil. Med. 2022, 46, 53–59. [Google Scholar] [CrossRef]
- Alzheimer’s Disease International. World Alzheimer Report 2021: Journey Through the Diagnosis of Dementia; Alzheimer’s Disease International: London, UK, 2021. [Google Scholar]
- Prince, M.; Albanese, E.; Guerchet, M.; Prina, M. Dementia and Risk Reduction: An Analysis of Protective and Modifiable Factors; Alzheimer’s Disease International: London, UK, 2014; pp. 1–104. [Google Scholar]
- Institute for Public Health (IPH). National Health and Morbidity Survey 2018: Elderly Health. Volume Two: Elderly Health Findings; Institute for Public Health (IPH): San Diego, CA, USA, 2019; Volume 2, ISBN 9788578110796. [Google Scholar]
- Ryan, N.S.; Nicholas, J.M.; Weston, P.S.J.; Liang, Y.; Lashley, T.; Guerreiro, R.; Adamson, G.; Kenny, J.; Beck, J.; Chavez-Gutierrez, L.; et al. Clinical Phenotype and Genetic Associations in Autosomal Dominant Familial Alzheimer’s Disease: A Case Series. Lancet Neurol. 2016, 15, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Valdez-Gaxiola, C.A.; Rosales-Leycegui, F.; Gaxiola-Rubio, A.; Moreno-Ortiz, J.M.; Figuera, L.E. Early- and Late-Onset Alzheimer’s Disease: Two Sides of the Same Coin? Diseases 2024, 12, 110. [Google Scholar] [CrossRef]
- Cho, Y.; Bae, H.G.; Okun, E.; Arumugam, T.V.; Jo, D.G. Physiology and Pharmacology of Amyloid Precursor Protein. Pharmacol. Ther. 2022, 235, 108122. [Google Scholar] [CrossRef] [PubMed]
- Vasques, J.F.; Heringer, P.V.B.; Gonçalves, R.G.d.J.; Campello-Costa, P.; Serfaty, C.A.; da Cunha Faria-Melibeu, A. Monocular Denervation of Visual Nuclei Modulates APP Processing and SAPPα Production: A Possible Role on Neural Plasticity. Int. J. Dev. Neurosci. 2017, 60, 16–25. [Google Scholar] [CrossRef] [PubMed]
- De Jonghe, C.; Esselens, C.; Kumar-Singh, S.; Craessaerts, K.; Serneels, S.; Checler, F.; Annaert, W.; Van Broeckhoven, C.; De Strooper, B. Pathogenic APP Mutations near the γ-Secretase Cleavage Site Differentially Affect Aβ Secretion and APP C-Terminal Fragment Stability. Hum. Mol. Genet. 2001, 10, 1665–1671. [Google Scholar] [CrossRef] [PubMed]
- Lanoiselée, H.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 Mutations in Early-Onset Alzheimer Disease: A Genetic Screening Study of Familial and Sporadic Cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed]
- Bagyinszky, E.; Youn, Y.C.; An, S.S.A.; Kim, S. The Genetics of Alzheimer’s Disease. Clin. Interv. Aging 2014, 9, 535–551. [Google Scholar] [CrossRef]
- Dai, M.H.; Zheng, H.; Zeng, L.D.; Zhang, Y. The genes associated with early-onset Alzheimer’s disease. Oncotarget 2017, 9, 15132–15143. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, E.H.; Ilagan, M.X.G.; Brunkan, A.L.; Hecimovic, S.; Li, Y.M.; Xu, M.; Lewis, H.D.; Saxena, M.T.; De Strooper, B.; Coonrod, A.; et al. A Presenilin Dimer at the Core of the γ-Secretase Enzyme: Insights from Parallel Analysis of Notch 1 and APP Proteolysis. Proc. Natl. Acad. Sci. USA 2003, 100, 13075–13080. [Google Scholar] [CrossRef]
- De Strooper, B.; Saftig, P.; Craessaerts, K.; Vanderstichele, H.; Guhde, G.; Annaert, W.; Von Figura, K.; Van Leuven, F. Deficiency of Presenilin-1 Inhibits the Normal Cleavage of Amyloid Precursor Protein. Nature 1998, 391, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Fraser, P.E.; Yang, D.S.; Yu, G.; Lévesque, L.; Nishimura, M.; Arawaka, S.; Serpell, L.C.; Rogaeva, E.; St George-Hyslop, P. Presenilin Structure, Function and Role in Alzheimer Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2000, 1502, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Cruts, M.; Theuns, J.; Van Broeckhoven, C. Locus-Specific Mutation Databases for Neurodegenerative Brain Diseases. Hum. Mutat. 2012, 33, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Lendon, C.; Mann, D.M.A.; Harris, J.M.; Chartier-Harlin, M.C.; Cumming, A.; Coates, J.; Lemmon, H.; StClair, D.; Iwatsubo, T. The −48 C/T Polymorphism in the Presenilin 1 Promoter Is Associated with an Increased Risk of Developing Alzheimer’s Disease and an Increased Aβ Load in Brain. J. Med. Genet. 2001, 38, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Tortosa, E.; Barquero, S.; Barón, M.; Gil-Neciga, E.; Castellanos, F.; Zurdo, M.; Manzano, S.; Muoz, D.G.; Jiménez-Huete, A.; Rábano, A.; et al. Clinical-Genetic Correlations in Familial Alzheimer’s Disease Caused by Presenilin 1 Mutations. J. Alzheimer’s Dis. 2010, 19, 873–884. [Google Scholar] [CrossRef]
- Pensalfini, A.; Albay, R.; Rasool, S.; Wu, J.W.; Hatami, A.; Arai, H.; Margol, L.; Milton, S.; Poon, W.W.; Corrada, M.M.; et al. Intracellular Amyloid and the Neuronal Origin of Alzheimer Neuritic Plaques. Neurobiol. Dis. 2014, 71, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Saura, C.A. Presenilin/γ-Secretase and Inflammation. Front. Aging Neurosci. 2010, 2, 1658. [Google Scholar] [CrossRef]
- Qin, J.; Zhang, X.; Wang, Z.; Li, J.; Zhang, Z.; Gao, L.; Ren, H.; Qian, M.; Du, B. Presenilin 2 Deficiency Facilitates Aβ-Induced Neuroinflammation and Injury by Upregulating P2X7 Expression. Sci. China Life Sci. 2017, 60, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Jayadev, S.; Leverenz, J.B.; Steinbart, E.; Stahl, J.; Klunk, W.; Yu, C.E.; Bird, T.D. Alzheimer’s Disease Phenotypes and Genotypes Associated with Mutations in Presenilin 2. Brain 2010, 133, 1143–1154. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; An, S.S.A.; Kim, S. Mutations in Presenilin 2 and Its Implications in Alzheimer’s Disease and Other Dementia-Associated Disorders. Clin. Interv. Aging 2015, 10, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Walker, E.S.; Martinez, M.; Brunkan, A.L.; Goate, A. Presenilin 2 Familial Alzheimer’s Disease Mutations Result in Partial Loss of Function and Dramatic Changes in Aβ 42/40 Ratios. J. Neurochem. 2005, 92, 294–301. [Google Scholar] [CrossRef]
- Morris, J.C.; Schindler, S.E.; McCue, L.M.; Moulder, K.L.; Benzinger, T.L.S.; Cruchaga, C.; Fagan, A.M.; Grant, E.; Gordon, B.A.; Holtzman, D.M.; et al. Assessment of Racial Disparities in Biomarkers for Alzheimer Disease. JAMA Neurol. 2019, 76, 264–273. [Google Scholar] [CrossRef]
- Shea, Y.F.; Chu, L.W.; Chan, A.O.K.; Ha, J.; Li, Y.; Song, Y.Q. A Systematic Review of Familial Alzheimer’s Disease: Differences in Presentation of Clinical Features among Three Mutated Genes and Potential Ethnic Differences. J. Formos. Med. Assoc. 2016, 115, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Tricco, A.C.; Lillie, E.; Zarin, W.; O’Brien, K.K.; Colquhoun, H.; Levac, D.; Moher, D.; Peters, M.D.J.; Horsley, T.; Weeks, L.; et al. PRISMA Extension for Scoping Reviews (PRISMA-ScR): Checklist and Explanation. Ann. Intern. Med. 2018, 169, 467–473. [Google Scholar] [CrossRef]
- Hsu, J.L.; Lin, C.H.; Chen, P.L.; Lin, K.J.; Chen, T.F. Genetic Study of Young-Onset Dementia Using Targeted Gene Panel Sequencing in Taiwan. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2021, 186, 67–76. [Google Scholar] [CrossRef]
- Liang, Z.; Wu, Y.; Li, C.; Liu, Z. Clinical and Genetic Characteristics in a Central-Southern Chinese Cohort of Early-Onset Alzheimer’s Disease. Front. Neurol. 2023, 14, 1119326. [Google Scholar] [CrossRef] [PubMed]
- Van Giau, V.; Bagyinszky, E.; Yang, Y.S.; Youn, Y.C.; An, S.S.A.; Kim, S.Y. Genetic Analyses of Early-Onset Alzheimer’s Disease Using next Generation Sequencing. Sci. Rep. 2019, 9, 8368. [Google Scholar] [CrossRef]
- Perrone, F.; Bjerke, M.; Hens, E.; Sieben, A.; Timmers, M.; De Roeck, A.; Vandenberghe, R.; Sleegers, K.; Martin, J.J.; De Deyn, P.P.; et al. Amyloid-Β1-43cerebrospinal Fluid Levels and the Interpretation of APP, PSEN1 and PSEN2 Mutations. Alzheimer’s Res. Ther. 2020, 12, 108. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.S.; Yang, Z.H.; Zhang, Y.; Yang, J.; Shang, D.D.; Zhang, S.Y.; Wu, J.; Ji, Y.; Zhao, L.; Xu, Y.M.; et al. Two Novel Mutations and a de Novo Mutation in PSEN1 in Early-Onset Alzheimer’s Disease. Aging Dis. 2019, 10, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Ren, R.J.; Zhong, Z.L.; Dammer, E.; Zhao, Q.H.; Shan, S.; Zhou, Z.; Li, X.; Zhang, Y.Q.; Cui, H.L.; et al. Mutation Profile of APP, PSEN1, and PSEN2 in Chinese Familial Alzheimer’s Disease. Neurobiol. Aging 2019, 77, 154–157. [Google Scholar] [CrossRef]
- Jiao, B.; Liu, H.; Guo, L.; Xiao, X.; Liao, X.; Zhou, Y.; Weng, L.; Zhou, L.; Wang, X.; Jiang, Y.; et al. The Role of Genetics in Neurodegenerative Dementia: A Large Cohort Study in South China. NPJ Genom. Med. 2021, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Shen, L.; Jia, L.; Wang, Q.; Li, F.; Li, Y.; Jia, J. A Novel PSEN1 M139L Mutation Found in a Chinese Pedigree with Early-Onset Alzheimer’s Disease Increases Aβ42 /Aβ40 Ratio. J. Alzheimer’s Dis. 2019, 69, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Quan, M.; Zhao, T.; Tang, Y.; Luo, P.; Wang, W.; Qin, Q.; Li, T.; Wang, Q.; Fang, J.; Jia, J. Effects of Gene Mutation and Disease Progression on Representative Neural Circuits in Familial Alzheimer’s Disease. Alzheimer’s Res. Ther. 2020, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Zhou, J.; Li, H.L.; Chen, Y.G.; Cheng, H.R.; Ye, L.Q.; Liu, D.S.; Chen, D.F.; Tao, Q.Q.; Wu, Z.Y. Mutation Screening in Chinese Patients with Familial Alzheimer’s Disease by Whole-Exome Sequencing. Neurobiol. Aging 2019, 76, 215.e15–215.e21. [Google Scholar] [CrossRef] [PubMed]
- Hyun, H.K.; Kim, Y.; Mi, P.Á. Presenilin 1 Gene Mutation (M139I) in a Patient with an Early-Onset Alzheimer’s Disease: Clinical Characteristics and Genetic Identification. Neurol. Sci. 2010, 31, 781–783. [Google Scholar] [CrossRef]
- Lacour, M.; Quenez, O.; Rovelet-Lecrux, A.; Salomon, B.; Rousseau, S.; Richard, A.C.; Quillard-Muraine, M.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. Causative Mutations and Genetic Risk Factors in Sporadic Early Onset Alzheimer’s Disease before 51 Years. J. Alzheimer’s Dis. 2019, 71, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Iragui, M.; Balasa, M.; Benejam, B.; Alcolea, D.; Fernández, S.; Videla, L.; Sala, I.; Sánchez-Saudinós, M.B.; Morenas-Rodriguez, E.; Ribosa-Nogué, R.; et al. Cerebral Amyloid Angiopathy in Down Syndrome and Sporadic and Autosomal-Dominant Alzheimer’s Disease. Alzheimer’s Dement. 2017, 13, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Alinaghi, S.; Tafakhori, A.; Sikora, E.; Azcona, L.J.; Karkheiran, S.; Goate, A.; Paisán-Ruiz, C.; Darvish, H. Genetic Screening in Two Iranian Families with Early-Onset Alzheimer’s Disease Identified a Novel PSEN1 Mutation. Neurobiol. Aging 2018, 62, 244.e15–244.e17. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.S.; Cheng, C.Y.; Liao, Y.C.; Hong, C.J.; Fuh, J.L. Mutational Analysis in Familial Alzheimer’s Disease of Han Chinese in Taiwan with a Predominant Mutation PSEN1 p.Met146Ile. Sci. Rep. 2020, 10, 19769. [Google Scholar] [CrossRef]
- Bartoletti-stella, A.; Tarozzi, M.; Mengozzi, G.; Asirelli, F.; Brancaleoni, L.; Mometto, N.; Stanzani-maserati, M.; Baiardi, S.; Ferriani, E.; Caffarra, P.; et al. Dementia-Related Genetic Variants in an Italian Population of Early-Onset Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 969817. [Google Scholar] [CrossRef]
- Wingo, T.S.; Cutler, D.J.; Wingo, A.P.; Le, N.A.; Rabinovici, G.D.; Miller, B.L.; Lah, J.J.; Levey, A.I. Association of Early-Onset Alzheimer Disease with Elevated Low-Density Lipoprotein Cholesterol Levels and Rare Genetic Coding Variants of APOB. JAMA Neurol. 2019, 76, 809–817. [Google Scholar] [CrossRef]
- Xiao, X.; Liu, H.; Liu, X.; Zhang, W.; Zhang, S.; Jiao, B. APP, PSEN1, and PSEN2 Variants in Alzheimer’s Disease: Systematic Re-Evaluation According to ACMG Guidelines. Front. Aging Neurosci. 2021, 13, 695808. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.H.; Seelaar, H.; Melhem, S.; Rozemuller, A.J.M.; van Swieten, J.C. Genetic Screening in Early-Onset Alzheimer’s Disease Identified Three Novel Presenilin Mutations. Neurobiol. Aging 2020, 86, 201.e9–201.e14. [Google Scholar] [CrossRef] [PubMed]
- Flood, D.G.; Reaume, A.G.; Dorfman, K.S.; Lin, Y.G.; Lang, D.M.; Trusko, S.P.; Savage, M.J.; Annaert, W.G.; De Strooper, B.; Siman, R.; et al. FAD Mutant PS-1 Gene-Targeted Mice: Increased Aβ42 and Aβ Deposition without APP Overproduction. Neurobiol. Aging 2002, 23, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Nosheny, R.L.; Insel, P.S.; Truran, D.; Schuff, N.; Jack, C.R.; Aisen, P.S.; Shaw, L.M.; Trojanowski, J.Q.; Weiner, M.W. Variables Associated with Hippocampal Atrophy Rate in Normal Aging and Mild Cognitive Impairment. Neurobiol. Aging 2015, 36, 273–282. [Google Scholar] [CrossRef]
- Shi, Z.; Wang, Y.; Liu, S.; Liu, M.; Liu, S.; Zhou, Y.; Wang, J.; Cai, L.; Huo, Y.R.; Gao, S.; et al. Clinical and Neuroimaging Characterization of Chinese Dementia Patients with Psen1 and Psen2 Mutations. Dement. Geriatr. Cogn. Disord. 2015, 39, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Van Giau, V.; Pyun, J.M.; Bagyinszky, E.; An, S.S.A.; Kim, S. A Pathogenic PSEN2 p.His169Asn Mutation Associated with Early-Onset Alzheimer’s Disease. Clin. Interv. Aging 2018, 13, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, J.; Shi, Y.; Wang, W.; Ren, Z.; Xia, M.; Zhang, Y.; Yang, M. Gene Mutations in a Han Chinese Alzheimer’s Disease Cohort. Brain Behav. 2019, 9, e01180. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.; Hardy, J. The Presenilins and Alzheimer’s Disease. Hum. Mol. Genet. 1997, 6, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Van Giau, V.; Bagyinszky, E.; Youn, Y.C.; An, S.S.A.; Kim, S.Y. APP, PSEN1, and PSEN2 Mutations in Asian Patients with Early-Onset Alzheimer Disease. Int. J. Mol. Sci. 2019, 20, 4757. [Google Scholar] [CrossRef] [PubMed]
- Dumois-Petersen, S.; Gallegos-Arreola, M.P.; Magaña-Torres, M.T.; Perea-Díaz, F.J.; Ringman, J.M.; Figuera, L.E. Autosomal Dominant Early Onset Alzheimer’s Disease in the Mexican State of Jalisco: High Frequency of the Mutation PSEN1 c.1292C>A and Phenotypic Profile of Patients. Am. J. Med. Genet. Part C Semin. Med. Genet. 2020, 184, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Willumsen, N.; Poole, T.; Nicholas, J.M.; Fox, N.C.; Ryan, N.S.; Lashley, T. Variability in the Type and Layer Distribution of Cortical Aβ Pathology in Familial Alzheimer’s Disease. Brain Pathol. 2021, 32, e13009. [Google Scholar] [CrossRef] [PubMed]
- Csaban, D.; Illes, A.; Renata, T.B.; Balicza, P.; Pentelenyi, K.; Molnar, V.; Gezsi, A.; Grosz, Z.; Gal, A.; Kovacs, T.; et al. Genetic landscape of early-onset dementia in Hungary. Neurol. Sci. 2022, 43, 5289–5300. [Google Scholar] [CrossRef] [PubMed]
- Petok, J.R.; Myers, C.E.; Pa, J.; Hobel, Z.; Wharton, D.M.; Medina, L.D.; Casado, M.; Coppola, G.; Gluck, M.A.; Ringman, J.M. Impairment of memory generalization in preclinical autosomal dominant Alzheimer’s disease mutation carriers. Neurobiol. Aging. 2018, 65, 149–157. [Google Scholar] [CrossRef]
- Soosman, S.K.; Joseph-Mathurin, N.; Braskie, M.N.; Bordelon, Y.M.; Wharton, D.; Casado, M.; Coppola, G.; McCallum, H.; Nuwer, M.; Coutin-Churchman, p.; et al. Widespread white matter and conduction defects in PSEN1-related spastic paraparesis. Neurobiol. Aging. 2016, 4, 201–209. [Google Scholar] [CrossRef]
- Quiroz, Y.T.; Zetterberg, H.; Reiman, E.M.; Chen, Y.; Su, Y.; Fox-Fuller, J.T.; Garcia, G.; Villegas, A.; Sepulveda-Falla, D.; Villada, M.; et al. Plasma neurofilament light chain in the presenilin 1 E280A autosomal dominant Alzheimer’s disease kindred: A cross-sectional and longitudinal cohort study. Lancet Neurol. 2020, 19, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.; Mozaffar, T.; Messmore, A.; Deignan, J.L.; Kimonis, V.E.; Ringman, J.M. Homozygosity for the A431E mutation in PSEN1 presenting with a relatively aggressive phenotype. Neurosci. Lett. 2019, 699, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Tariot, P.N.; Lopera, F.; Langbaum, J.B.; Thomas, R.G.; Hendrix, S.; Schneider, L.S.; Rios-Romenets, S.; Giraldo, M.; Acosta, N.; Tobon, C.; et al. Alzheimer’s Prevention Initiative. The Alzheimer’s Prevention Initiative Autosomal-Dominant Alzheimer’s Disease Trial: A study of crenezumab versus placebo in preclinical PSEN1 E280A mutation carriers to evaluate efficacy and safety in the treatment of autosomal-dominant Alzheimer’s disease, including a placebo-treated noncarrier cohort. Alzheimers Dement. 2018, 4, 150–160. [Google Scholar] [CrossRef]
- Luukkainen, L.; Helisalmi, S.; Kytövuori, L.; Ahmasalo, R.; Solje, E.; Haapasalo, A.; Hiltunen, M.; Remes, A.M.; Krüger, J. Mutation Analysis of the Genes Linked to Early Onset Alzheimer’s Disease and Frontotemporal Lobar Degeneration. J. Alzheimer’s Dis. 2019, 69, 775–782. [Google Scholar] [CrossRef] [PubMed]
- N’Songo, A.; Carrasquillo, M.M.; Wang, X.; Nguyen, T.; Asmann, Y.; Younkin, S.G.; Allen, M.; Duara, R.; Custo, M.T.; Graff-Radford, N.; et al. Comprehensive Screening for Disease Risk Variants in Early-Onset Alzheimer’s Disease Genes in African Americans Identifies Novel PSEN Variants. J. Alzheimers Dis. 2017, 56, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Abdala, B.B.; Dos Santos, J.M.; Gonçalves, A.P.; da Motta, L.B.; Laks, J.; de Borges, M.B.; Gonçalves Pimentel, M.M.; Santos-Rebouças, C.B. Influence of low frequency PSEN1 variants on familial Alzheimer’s disease risk in Brazil. Neurosci. Lett. 2017, 653, 341–345. [Google Scholar] [CrossRef]
- Ramos-Campoy, O.; Antonell, A.; Falgàs, N.; Balasa, M.; Borrego-Écija, S.; Rodríguez-Santiago, B.; Datta, D.; Armengol, L.; Fernández-Villullas, G.; Bosch, B.; et al. Screening of dementia genes by whole-exome sequencing in Spanish patients with early-onset dementia: Likely pathogenic, uncertain significance and risk variants. Neurobiol. Aging 2020, 93, e1–e9. [Google Scholar] [CrossRef] [PubMed]
- de la Vega, M.P.; Näslund, C.; Brundin, R.; Lannfelt, L.; Löwenmark, M.; Kilander, L.; Ingelsson, M.; Giedraitis, V. Mutation analysis of disease causing genes in patients with early onset or familial forms of Alzheimer’s disease and frontotemporal dementia. BMC Genom. 2022, 23, 99. [Google Scholar] [CrossRef]
- Perrone, F.; Cacace, R.; Van Mossevelde, S.; Van den Bossche, T.; De Deyn, P.P.; Cras, P.; Engelborghs, S.; van der Zee, J.; Van Broeckhoven, C. Genetic screening in early-onset dementia patients with unclear phenotype: Relevance for clinical diagnosis. Neurobiol. Aging. 2018, 69, e7–e292. [Google Scholar] [CrossRef]
- Bartoletti-Stella, A.; Baiardi, S.; Stanzani-Maserati, M.; Piras, S.; Caffarra, P.; Raggi, A.; Pantieri, R.; Baldassari, S.; Caporali, L.; Abu-Rumeileh, S.; et al. Identification of rare genetic variants in Italian patients with dementia by targeted gene sequencing. Neurobiol. Aging 2018, 66, e23–e180. [Google Scholar] [CrossRef] [PubMed]
- Takada, L.T. Genetic investigation of dementias in clinical practice. Arq. Neuropsiquiatr. 2022, 80 (Suppl. 1), 36–41. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Fu, Y.; Shen, L.; Zhang, H.; Zhu, M.; Qiu, Q.; Wang, Q.; Yan, X.; Kong, C.; Hao, J.; et al. PSEN1, PSEN2, and APP mutations in 404 Chinese pedigrees with familial Alzheimer’s disease. Alzheimers Dement. 2020, 16, 178–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Xie, Y.; Wang, W.; Feng, X.; Jia, J. Clinical characterization of an APP mutation (V717I) in five Han Chinese families with early-onset Alzheimer’s disease. J. Neurol. Sci. 2017, 372, 379–386. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, Y.; Meng, F.; Zhang, K.; Liu, X.; Peng, G. Presenilin 1 and APP Gene Mutations in Early-Onset AD Families from a Southeast Region of China. Curr. Alzheimer Res. 2020, 17, 540–546. [Google Scholar] [CrossRef]
- An, S.S.; Park, S.A.; Bagyinszky, E.; Bae, S.O.; Kim, Y.-J.; Im, J.Y.; Park, K.W.; Park, K.H.; Kim, E.-J.; Jeong, J.H.; et al. A genetic screen of the mutations in the Korean patients with early-onset Alzheimer’s disease. Clin. Interv. Aging 2016, 11, 1817–1822. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.Y.; Zhao, Q.H.; Huang, Q.; Dammer, E.; Chen, S.D.; Ren, R.J.; Wang, G.; Alzheimer’s Disease Neuroimaging Initiative. Genetic profiles of familial late-onset Alzheimer’s disease in China: The Shanghai FLOAD study. Genes Dis. 2021, 9, 1639–1649. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Characteristics | Number of Studies (n) | Percentage (%) |
|---|---|---|
| Countries | ||
| Asian | 36 | 47.4 |
| Western | 40 | 52.6 |
| Type of Study | ||
| Asian | ||
| Case Report | 11 | 30.6 |
| Brief Communication | 3 | 8.3 |
| Case–Control | 4 | 11.1 |
| Cohort | 6 | 16.7 |
| Cross-Sectional | 12 | 33.3 |
| Western | ||
| Case Report | 8 | 20 |
| Brief Communication | 0 | 0 |
| Case–Control | 3 | 7.5 |
| Cohort | 10 | 25 |
| Cross-Sectional | 19 | 47.5 |
| EOAD History | ||
| Asian | ||
| Familial | 27 | 75 |
| Sporadic | 1 | 2.8 |
| Familial and Sporadic | 6 | 16.7 |
| Unknown | 2 | 5.5 |
| Western | ||
| Familial | 26 | 65 |
| Sporadic | 3 | 7.5 |
| Familial and Sporadic | 9 | 22.5 |
| Unknown | 2 | 5 |
| Mutations Mutually Present in Asian and Western Populations | Clinical Phenotype (Asian) | Clinical Phenotype (Western) |
|---|---|---|
| G206D | memory decline, mental and behavioral change | isolated progressive cognitive decline |
| G206S | earlier mean AAO, severe cognitive impairment | memory impairment, disorientation, hallucination, psychotic sessions, conversion, mixed dissociative disorder, myoclonus, impaired speech, and apraxia |
| G206V | slowly progressing memory loss combined with irritation and anxiety | myoclonus, seizures |
| M139V | memory decline, language impairment, mental and behavioral change | behavioral presentation, myoclonus, seizures, spastic paraparesis with/without other pyramidal signs |
| M146I | myoclonus, seizure, extrapyramidal symptoms, emotional lability | isolated progressive cognitive decline |
| M146V | memory decline, language impairment, mental and behavioral change | Slight short-term memory dysfunction and spatial disorientation, myoclonic epileptic seizures, and gait difficulties. |
| V717I | memory decline, language impairment, mental and behavioral change | typical of AD with amnestic presentation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poniah, P.; Abdul Rashed, A.; Abdul Jalil, J.; Ali, E.Z. Clinical Significance of Early-Onset Alzheimer’s Mutations in Asian and Western Populations: A Scoping Review. Genes 2025, 16, 345. https://doi.org/10.3390/genes16030345
Poniah P, Abdul Rashed A, Abdul Jalil J, Ali EZ. Clinical Significance of Early-Onset Alzheimer’s Mutations in Asian and Western Populations: A Scoping Review. Genes. 2025; 16(3):345. https://doi.org/10.3390/genes16030345
Chicago/Turabian StylePoniah, Prevathe, Aswir Abdul Rashed, Julaina Abdul Jalil, and Ernie Zuraida Ali. 2025. "Clinical Significance of Early-Onset Alzheimer’s Mutations in Asian and Western Populations: A Scoping Review" Genes 16, no. 3: 345. https://doi.org/10.3390/genes16030345
APA StylePoniah, P., Abdul Rashed, A., Abdul Jalil, J., & Ali, E. Z. (2025). Clinical Significance of Early-Onset Alzheimer’s Mutations in Asian and Western Populations: A Scoping Review. Genes, 16(3), 345. https://doi.org/10.3390/genes16030345