
Transcriptomic Profiling Highlights Metabolic and Biosynthetic Pathways Involved in In Vitro Flowering in Anoectochilus roxburghii (Wall.) Lindl.

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Library Construction and Transcriptome Sequencing
2.3. Differential Gene Expression Analysis
2.4. Annotation
2.5. PPI Network
2.6. Statistical Analyses
3. Results
3.1. Gene Expression Analysis
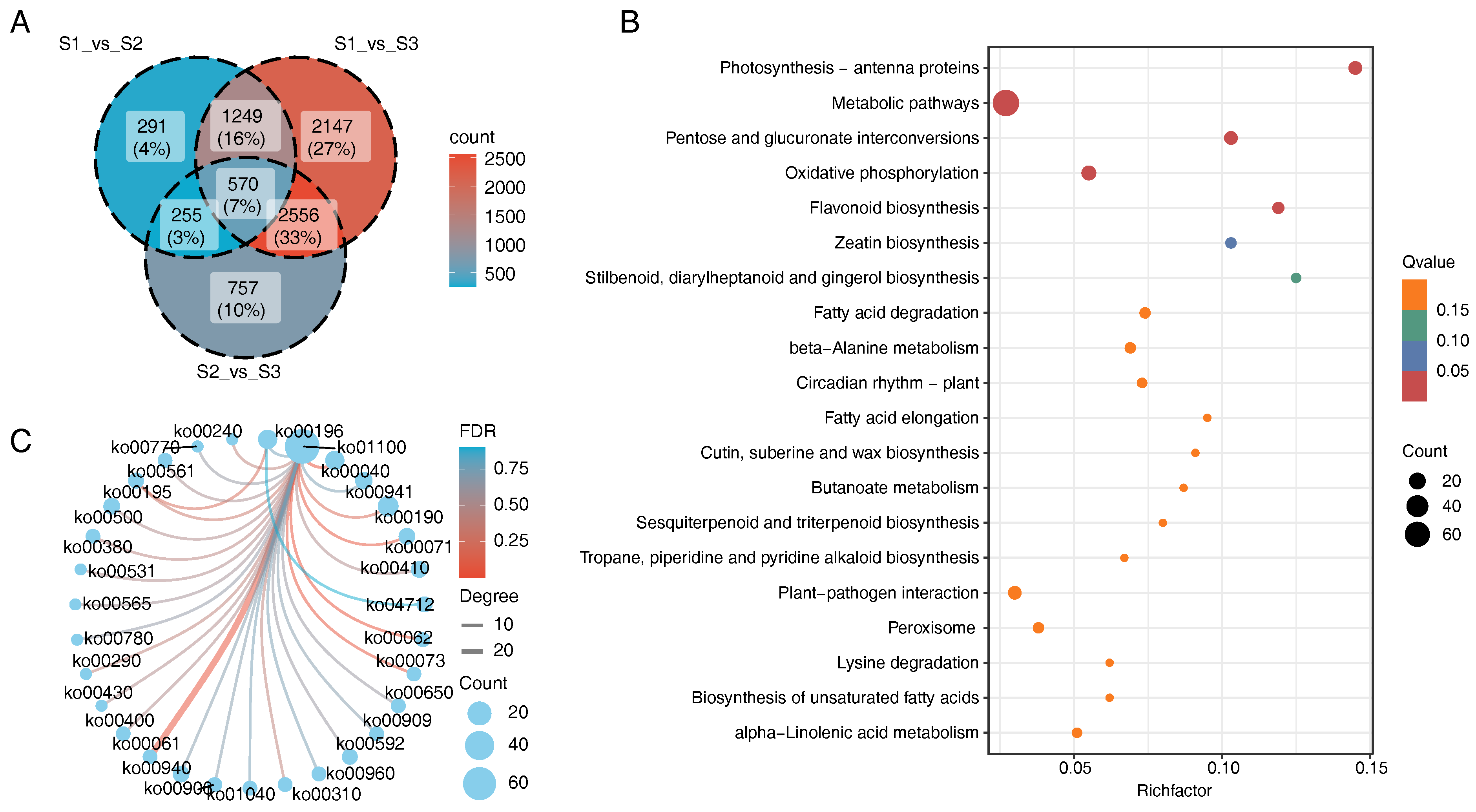
3.2. Differentially Expressed Gene (DEG) Screening
3.3. Trend Analysis
3.4. Enrichment Analysis
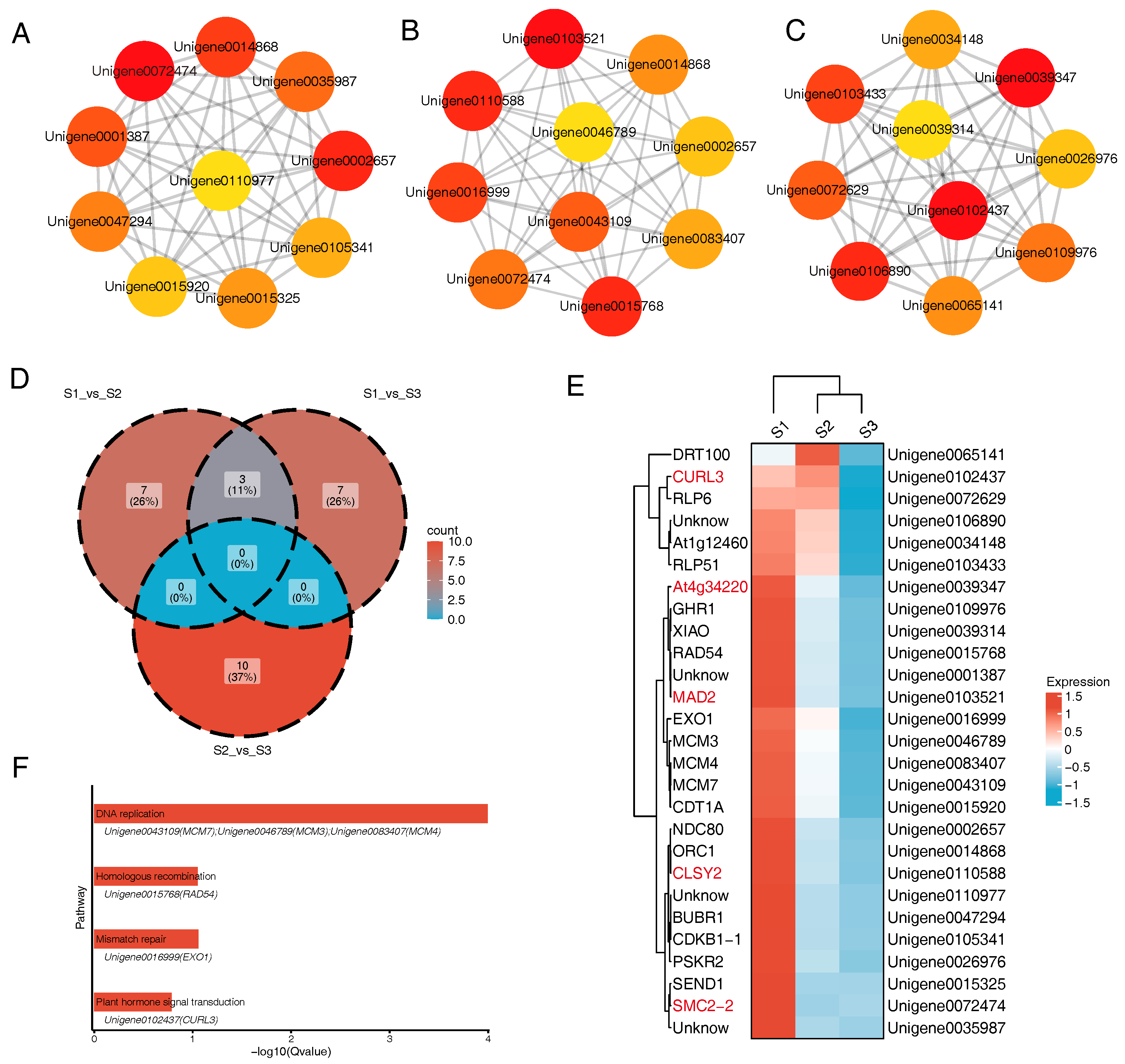
3.5. Core Gene Analysis
3.6. PPI Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hong, L.; Shao, Q.S.; Zhou, A.C.; Wang, H.Z.; Zhang, A.L.; Xu, J.W.; Huang, Y.Q. Current status and sustainable development countermeasures of Anoectochilus Roxburghii. China J. Chin. Mater. Med. 2016, 41, 553–558. [Google Scholar] [CrossRef]
- Shao, Q.; Deng, Y.; Liu, H.; Zhang, A.; Huang, Y.; Xu, G.; Li, M. Essential oils extraction from Anoectochilus Roxburghii using supercritical carbon dioxide and their antioxidant activity. Ind. Crop Prod. 2014, 60, 104–112. [Google Scholar] [CrossRef]
- Xu, M.; Shao, Q.; Ye, S.; Li, S.; Wu, M.; Ding, M.; Li, Y. Simultaneous Extraction and Identification of Phenolic Compounds in Anoectochilus roxburghii Using Microwave-Assisted Extraction Combined with UPLC-Q-TOF-MS/MS and Their Antioxidant Activities. Front. Plant Sci. 2017, 8, 1474. [Google Scholar] [CrossRef]
- Ito, A.; Kasai, R.; Yamasaki, K.; Sugimoto, H. Aliphatic and aromatic glucosides from Anoectochilus koshunensis. Phytochemistry 1993, 33, 1133–1137. [Google Scholar] [CrossRef]
- Zhang, A.; Wang, H.; Shao, Q.; Xu, M.; Zhang, W.; Li, M. Large scale in vitro propagation of Anoectochilus roxburghii for commercial application: Pharmaceutically important and ornamental plant. Ind. Crop Prod. 2015, 70, 158–162. [Google Scholar] [CrossRef]
- Sherif, N.A.; Kumar, T.S.; Rao, M.V. In vitro regeneration by callus culture of Anoectochilus elatus Lindley, an endangered terrestrial jewel orchid. In Vitro Cell. Dev. Biol.-Plant 2016, 52, 72–80. [Google Scholar] [CrossRef]
- Murashige, T.; Skoog, F. A Revised Medium for Rapid Growth and Bioassays with Tobacco Tissue Cultures. Physiol. Plant. 1962, 15, 473–497. [Google Scholar] [CrossRef]
- Knudson, L. Flower Production by Orchid Grown Non-Symbiotically. Bot. Gaz. 1930, 89, 192. [Google Scholar] [CrossRef]
- Teixeira da Silva, J.A.; Kerbauy, G.B.; Zeng, S.; Chen, Z.; Duan, J. In vitro flowering of orchids. Crit. Rev. Biotechnol. 2014, 34, 56–76. [Google Scholar] [CrossRef] [PubMed]
- Murthy, K.S.R.; Kondamudi, R.; Chalapathi Rao, P.V.; Pullaiah, T. In Vitro Flowering—A Review. J. Agric. Technol. 2012, 8, 1517–1536. [Google Scholar]
- Wang, G.; XU, Z. In Vitro Flowering of Dendrobium candidum. Sci. China 1997, 40, 36–41. [Google Scholar]
- Wang, Z.H.; Wang, L.; Ye, Q.S. High frequency early flowering from in vitro seedlings of Dendrobium nobile. Sci. Hortic. 2009, 122, 328–331. [Google Scholar] [CrossRef]
- Chandler, J.W. The Hormonal Regulation of Flower Development. J. Plant Growth Regul. 2011, 30, 242–254. [Google Scholar] [CrossRef]
- Claeys, H.; De Bodt, S.; Inzé, D. Gibberellins and DELLAs: Central nodes in growth regulatory networks. Trends Plant Sci. 2014, 19, 231–239. [Google Scholar] [CrossRef]
- Mazzoni-Putman, S.M.; Brumos, J.; Zhao, C.; Alonso, J.M.; Stepanova, A.N. Auxin Interactions with Other Hormones in Plant Development. Cold Spring Harbor Perspect. Biol. 2021, 13, a039990. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, H. CYTOKININS: Activity, Biosynthesis, and Translocation. Annu. Rev. Plant Biol. 2006, 57, 431–449. [Google Scholar] [CrossRef]
- Blanchard, M.G.; Runkle, E.S. Benzyladenine Promotes Flowering in Doritaenopsis and Phalaenopsis Orchids. J. Plant Growth Regul. 2008, 27, 141–150. [Google Scholar] [CrossRef]
- Goh, C.J.; Yang, A.L. Effects of growth regulators and decapitation on flowering of Dendrobium orchid hybrids. Plant Sci. Lett. 1978, 12, 287–292. [Google Scholar] [CrossRef]
- Tan, F.C.; Swain, S.M. Genetics of flower initiation and development in annual and perennial plants. Physiol. Plant. 2006, 128, 8–17. [Google Scholar] [CrossRef]
- Capovilla, G.; Schmid, M.; Posé, D. Control of flowering by ambient temperature. J. Exp. Bot. 2015, 66, 59–69. [Google Scholar] [CrossRef]
- Zhou, Y.; Sun, X.D.; Ni, M. Timing of Photoperiodic Flowering: Light Perception and Circadian Clock. J. Integr. Plant Biol. 2007, 49, 28–34. [Google Scholar] [CrossRef]
- Yu, Q.; Jin, X.; Liu, C.; Wen, Y. An Integrated Analysis of Transcriptome and miRNA Sequencing Provides Insights into the Dynamic Regulations during Flower Morphogenesis in Petunia. Horticulturae 2022, 8, 284. [Google Scholar] [CrossRef]
- Fan, Y.; Zheng, Y.; Chen, L.; Xu, L.; Teixeira da Silva, J.A.; Wu, B.; Yu, X. Transcriptomic and proteomic analyses reveal changes in the metabolic pathways of Paeonia lactiflora petaloid stamens. Sci. Hortic. 2023, 312, 111859. [Google Scholar] [CrossRef]
- Jia, S.; Wang, Y.; Hu, J.; Ding, Z.; Liang, Q.; Zhang, Y.; Wang, H. Mineral and metabolic profiles in tea leaves and flowers during flower development. Plant Physiol. Biochem. 2016, 106, 316–326. [Google Scholar] [CrossRef]
- Sheldon, C.C.; Burn, J.E.; Perez, P.P.; Metzger, J.; Edwards, J.A.; Peacock, W.J.; Dennis, E.S. The FLF MADS Box Gene: A Repressor of Flowering in Arabidopsis Regulated by Vernalization and Methylation. Plant Cell 1999, 11, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yoo, S.J.; Park, S.H.; Hwang, I.; Lee, J.S.; Ahn, J.H. Role of SVP in the control of flowering time by ambient temperature in Arabidopsis. Genes Dev. 2007, 21, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Coen, E.S.; Meyerowitz, E.M. The war of the whorls: Genetic interactions controlling flower development. Nature 1991, 353, 31–37. [Google Scholar] [CrossRef]
- Teixeira da Silva, J.A.; Aceto, S.; Liu, W.; Yu, H.; Kanno, A. Genetic Control of flower development, color and senescence of Dendrobium orchids. Sci. Hortic. 2014, 175, 74–86. [Google Scholar] [CrossRef]
- Cantone, C.; Gaudio, L.; Aceto, S. The PI/GLO-like locus in Orchids: Duplication and purifying selection at synonymous sites within Orchidinae (Orchidaceae). Gene 2011, 481, 48–55. [Google Scholar] [CrossRef]
- Aceto, S.; Gaudio, L. The MADS and the Beauty: Genes Involved in the Development of Orchid Flowers. Curr. Genom. 2011, 12, 342–356. [Google Scholar] [CrossRef]
- Zhang, D.; Zhao, X.W.; Li, Y.Y.; Ke, S.J.; Yin, W.L.; Lan, S.; Liu, Z.J. Advances and prospects of orchid research and industrialization. Hortic. Res. 2022, 9, uhac220. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING V10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef] [PubMed]
- Otasek, D.; Morris, J.H.; Bouças, J.; Pico, A.R.; Demchak, B. Cytoscape Automation: Empowering workflow-based network analysis. Genome Biol. 2019, 20, 185. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. 2024. Available online: https://www.r-project.org/ (accessed on 10 December 2024).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef]
- Zhang, J. ClusterGVis: One-step to Cluster and Visualize Gene Expression Matrix. 2022. Available online: https://github.com/junjunlab/ClusterGVis (accessed on 10 December 2024).
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Gao, C.H.; Chen, C.; Akyol, T.; Dusa, A.; Yu, G.; Cao, B.; Cai, P. ggVennDiagram: Intuitive Venn siagram software extended. iMeta 2024, 3, e177. [Google Scholar] [CrossRef] [PubMed]
- Csárdi, G.; Nepusz, T.; Müller, K.; Horvát, S.; Traag, V.; Zanini, F.; Noom, D. Igraph for R: R Interface of the Igraph Library for Graph Theory and Network Analysis (v2.1.3). Zenodo 2025. [Google Scholar] [CrossRef]
- Pedersen, T.L. ggraph: An Implementation of Grammar of Graphics for Graphs and Networks. 2024. Available online: https://CRAN.R-project.org/package=ggraph (accessed on 10 December 2024).
- Wahl, V.; Ponnu, J.; Schlereth, A.; Arrivault, S.; Langenecker, T.; Franke, A.; Feil, R.; Lunn, J.E.; Stitt, M.; Schmid, M. Regulation of Flowering by Trehalose-6-Phosphate Signaling in Arabidopsis thaliana. Science 2013, 339, 704–707. [Google Scholar] [CrossRef]
- He, C.; Liu, X.; Teixeira da Silva, J.A.; Liu, N.; Zhang, M.; Duan, J. Transcriptome sequencing and metabolite profiling analyses provide comprehensive insight into molecular mechanisms of flower development in Dendrobium officinale (Orchidaceae). Plant Mol. Biol. 2020, 104, 529–548. [Google Scholar] [CrossRef]
- Xing, L.; Zhang, D.; Qi, S.; Chen, X.; An, N.; Li, Y.; Zhao, C.; Han, M.; Zhao, J. Transcription profiles reveal the regulatory mechanisms of spur bud changes and flower induction in response to shoot bending in apple (Malus domestica Borkh.). Plant Mol. Biol. 2019, 99, 45–66. [Google Scholar] [CrossRef] [PubMed]
- Jing, D.; Chen, W.; Hu, R.; Zhang, Y.; Xia, Y.; Wang, S.; He, Q.; Guo, Q.; Liang, G. An Integrative Analysis of Transcriptome, Proteome and Hormones Reveals Key Differentially Expressed Genes and Metabolic Pathways Involved in Flower Development in Loquat. Int. J. Mol. Sci. 2020, 21, 5107. [Google Scholar] [CrossRef]
- Zhou, X.; Shi, F.; Zhou, L.; Zhou, Y.; Liu, Z.; Ji, R.; Feng, H. iTRAQ-based proteomic analysis of fertile and sterile flower buds from a genetic male sterile line ‘AB01’ in Chinese cabbage (Brassica campestris L. ssp. Pekinensis). J. Proteom. 2019, 204, 103395. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Shi, X.; Zou, Q.; Tian, D.; An, X.; Zhu, K. iTRAQ-based quantitative proteomic analysis reveals dynamic changes during daylily flower senescence. Planta 2018, 248, 859–873. [Google Scholar] [CrossRef]
- Chakraborty, A.; Chaudhury, R.; Dutta, S.; Basak, M.; Dey, S.; Schäffner, A.R.; Das, M. Role of metabolites in flower development and discovery of compounds controlling flowering time. Plant Physiol. Biochem. 2022, 190, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Z.; Jin, W.; Fang, Y.; Yang, Q.; Xiang, J. Transcriptome analysis and identification of genes associated with flower development in Rhododendron pulchrum Sweet (Ericaceae). Gene 2018, 679, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Huang, S.; Liu, Y.; Xiao, F.; Liu, J.; Zhang, Z.; Chen, Q.; Mao, Y.; Cao, X.; Wang, Y.; et al. The transcriptome analyses of Tagetes erecta provides novel insights into secondary metabolite biosynthesis during flower development. Gene 2018, 660, 18–27. [Google Scholar] [CrossRef]
- Singh, A.; Breja, P.; Khurana, J.P.; Khurana, P. Wheat Brassinosteroid-Insensitive1 (TaBRI1) Interacts with Members of TaSERK Gene Family and Cause Early Flowering and Seed Yield Enhancement in Arabidopsis. PLoS ONE 2016, 11, e0153273. [Google Scholar] [CrossRef] [PubMed]
- Mumtaz, M.A.; Wang, Y.; Li, F.; Shang, L.; Wang, Y.; Zhang, X.; Tao, J.; Gai, W.; Dong, H.; Ahiakpa, J.K.; et al. Hindered tomato reproductive development by altered brassinosteroid sensitivity1 mutant. J. Plant Growth Regul. 2022, 96, 473–481. [Google Scholar] [CrossRef]
- Stevens, R.; Mariconti, L.; Rossignol, P.; Perennes, C.; Cella, R.; Bergounioux, C. Two E2F Sites in the Arabidopsis MCM3 Promoter Have Different Roles in Cell Cycle Activation and Meristematic Expression. J. Biol. Chem. 2002, 277, 32978–32984. [Google Scholar] [CrossRef] [PubMed]
- Bastida, M.; Puigdomènech, P. Specific expression of ZmPRL, the maize homolog of MCM7, during early embryogenesis. Plant Sci. 2002, 162, 97–106. [Google Scholar] [CrossRef]
- Dresselhaus, T.; Srilunchang, K.o.; Leljak-Levanić, D.; Schreiber, D.N.; Garg, P. The Fertilization-Induced DNA Replication Factor MCM6 of Maize Shuttles between Cytoplasm and Nucleus, and Is Essential for Plant Growth and Development. Plant Physiol. 2006, 140, 512–527. [Google Scholar] [CrossRef] [PubMed]
- Jacqmard, A.; Houssa, C. DNA fiber replication during a morphogenetic switch in the shoot meristematic cells of a higher plant. Exp. Cell Res. 1988, 179, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Durdan, S.F.; Herbert, R.J.; Francis, D. Activation of latent origins of DNA replication in florally determined shoot meristems of long-day and short-day plants: Silene coeli-rosa and Pharbitis nil. Planta 1998, 207, 235–240. [Google Scholar] [CrossRef]
- Fu, S.; Yang, Y.; Wang, P.; Ying, Z.; Xu, W.; Zhou, Z. Comparative transcriptomic analysis of normal and abnormal in vitro flowers in Cymbidium nanulum Y. S. Wu et S. C. Chen Identifies differentially expressed genes and candidate genes involved in flower formation. Front. Plant Sci. 2022, 13, 1007913. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Lu, C.; Gao, J.; Wei, Y.; Xie, Q.; Jin, J.; Zhu, G.; Yang, F. Integrated proteomic, transcriptomic, and metabolomic profiling reveals that the gibberellin–abscisic acid hub runs flower development in the Chinese orchid Cymbidium sinense. Hortic. Res. 2024, 11, uhae073. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, S.; Liu, J.; Cai, C.; Zhang, Y.; Fu, S.; Yang, Y.; Zhou, Z.; Ying, Z. Transcriptomic Profiling Highlights Metabolic and Biosynthetic Pathways Involved in In Vitro Flowering in Anoectochilus roxburghii (Wall.) Lindl. Genes 2025, 16, 132. https://doi.org/10.3390/genes16020132
Yu S, Liu J, Cai C, Zhang Y, Fu S, Yang Y, Zhou Z, Ying Z. Transcriptomic Profiling Highlights Metabolic and Biosynthetic Pathways Involved in In Vitro Flowering in Anoectochilus roxburghii (Wall.) Lindl. Genes. 2025; 16(2):132. https://doi.org/10.3390/genes16020132
Chicago/Turabian StyleYu, Shuisheng, Julian Liu, Chenchen Cai, Yi Zhang, Shuangbin Fu, Yanping Yang, Zhuang Zhou, and Zhen Ying. 2025. "Transcriptomic Profiling Highlights Metabolic and Biosynthetic Pathways Involved in In Vitro Flowering in Anoectochilus roxburghii (Wall.) Lindl." Genes 16, no. 2: 132. https://doi.org/10.3390/genes16020132
APA StyleYu, S., Liu, J., Cai, C., Zhang, Y., Fu, S., Yang, Y., Zhou, Z., & Ying, Z. (2025). Transcriptomic Profiling Highlights Metabolic and Biosynthetic Pathways Involved in In Vitro Flowering in Anoectochilus roxburghii (Wall.) Lindl. Genes, 16(2), 132. https://doi.org/10.3390/genes16020132