

Differential microRNA and Target Gene Expression in Scots Pine (Pinus sylvestris L.) Needles in Response to Methyl Jasmonate Treatment


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material and Extraction of RNA
2.2. Small RNA Enrichment, Library Preparation and Sequencing Analysis
2.3. Transcriptome Library Preparation and Sequencing Analysis
2.4. Data Analysis
2.5. Statistical Analysis
3. Results
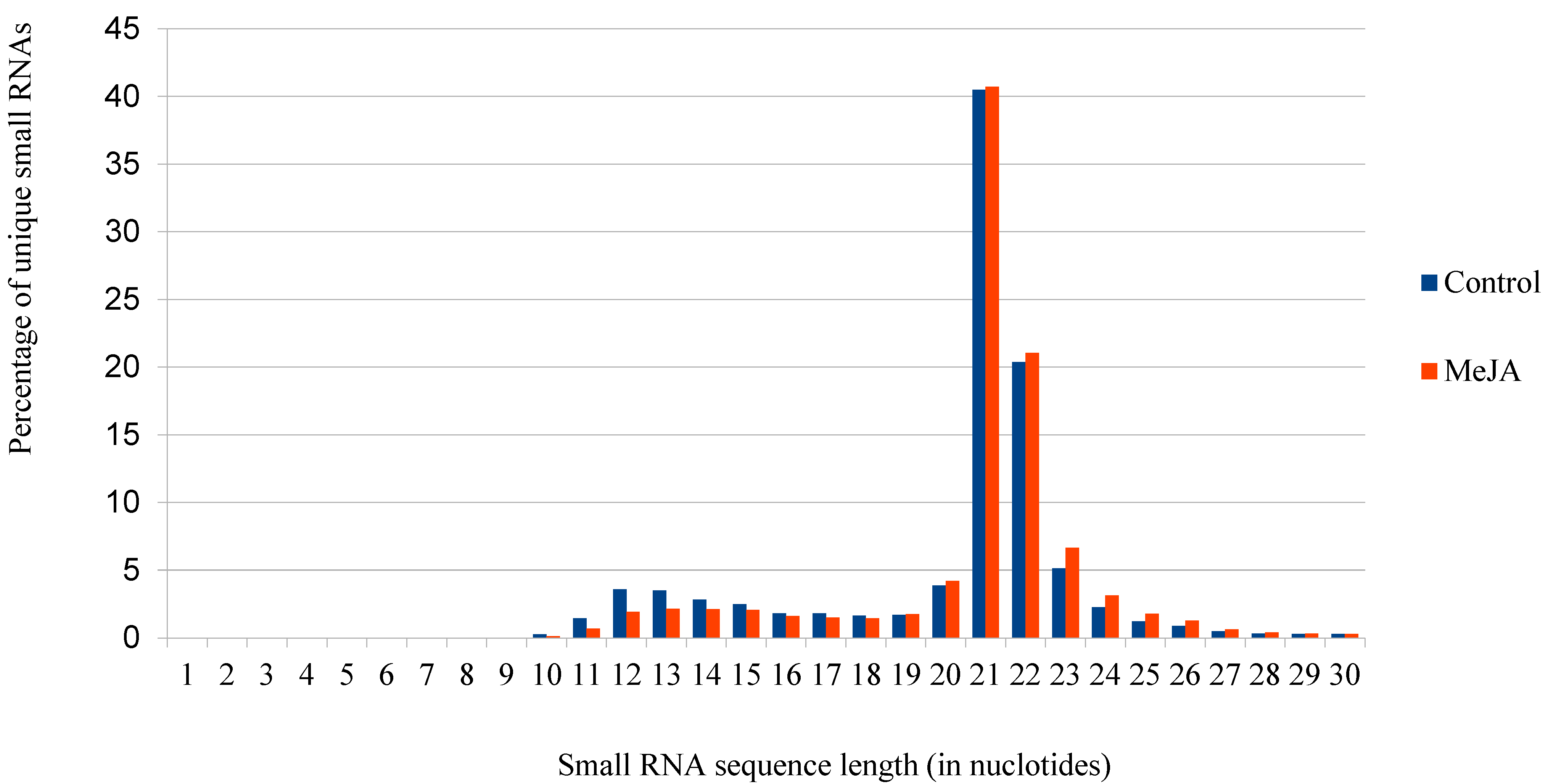
3.1. Sequencing of Small RNA Libraries
3.2. Differentially Expressed microRNAs
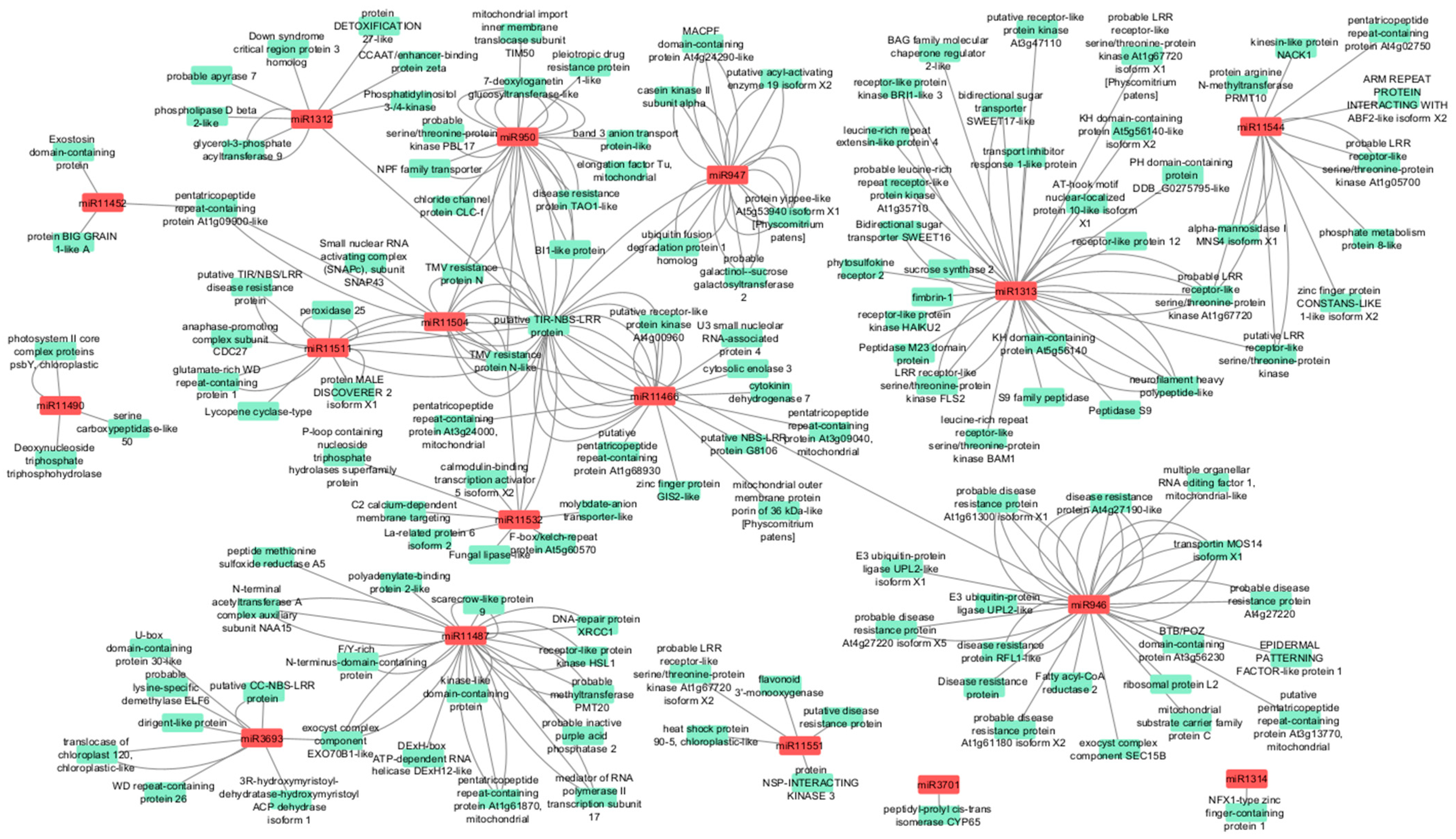
3.3. Gene Ontology Annotation of Target Genes of Differentially Expressed microRNAs
3.4. Target Genes of Differentially Expressed Conifer-Specific microRNAs
3.5. Identification and Expression Analysis of Target Genes of Differentially Expressed microRNAs from Transcriptome Data
3.6. Resistance-Related Target Genes of Differentially Expressed microRNAs from Transcriptome Data
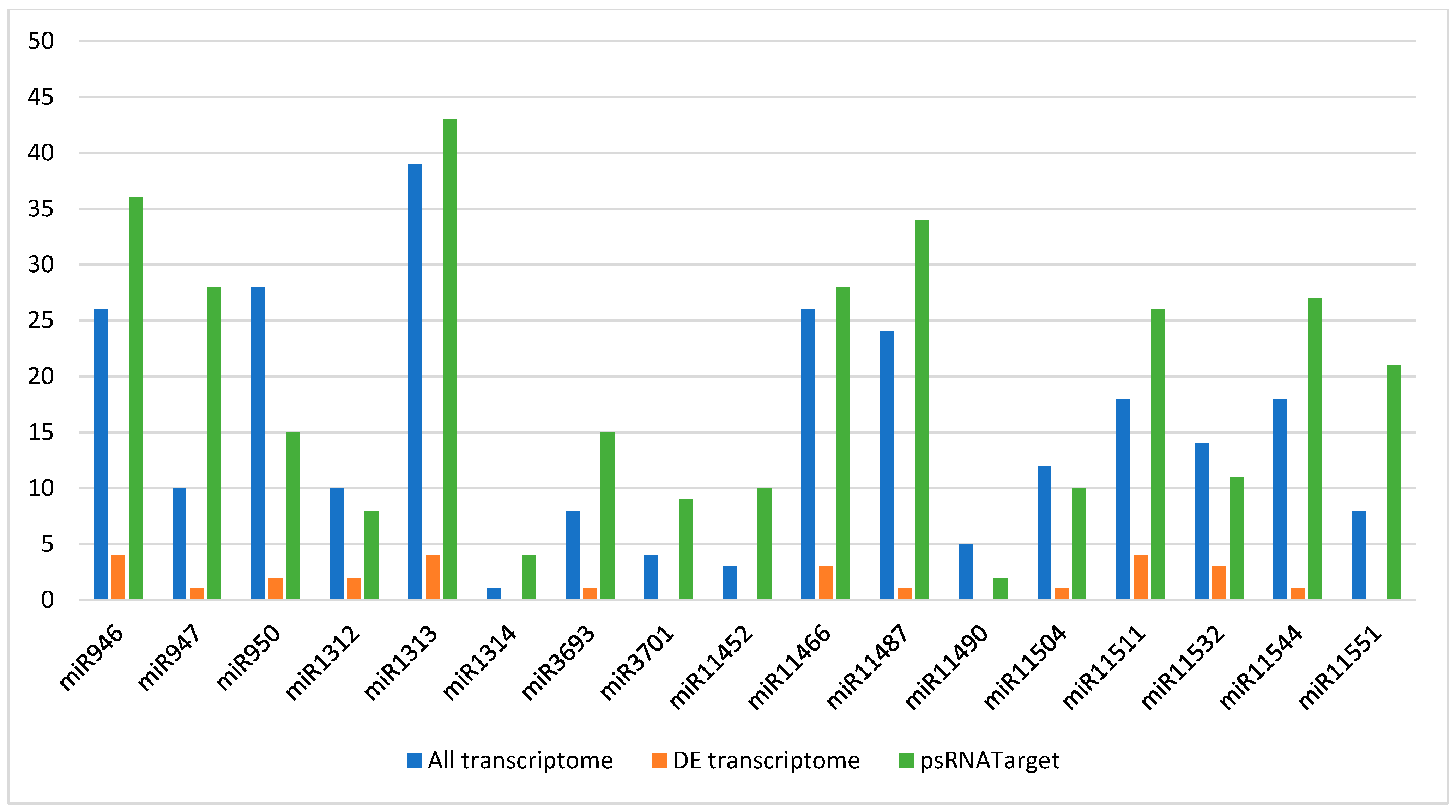
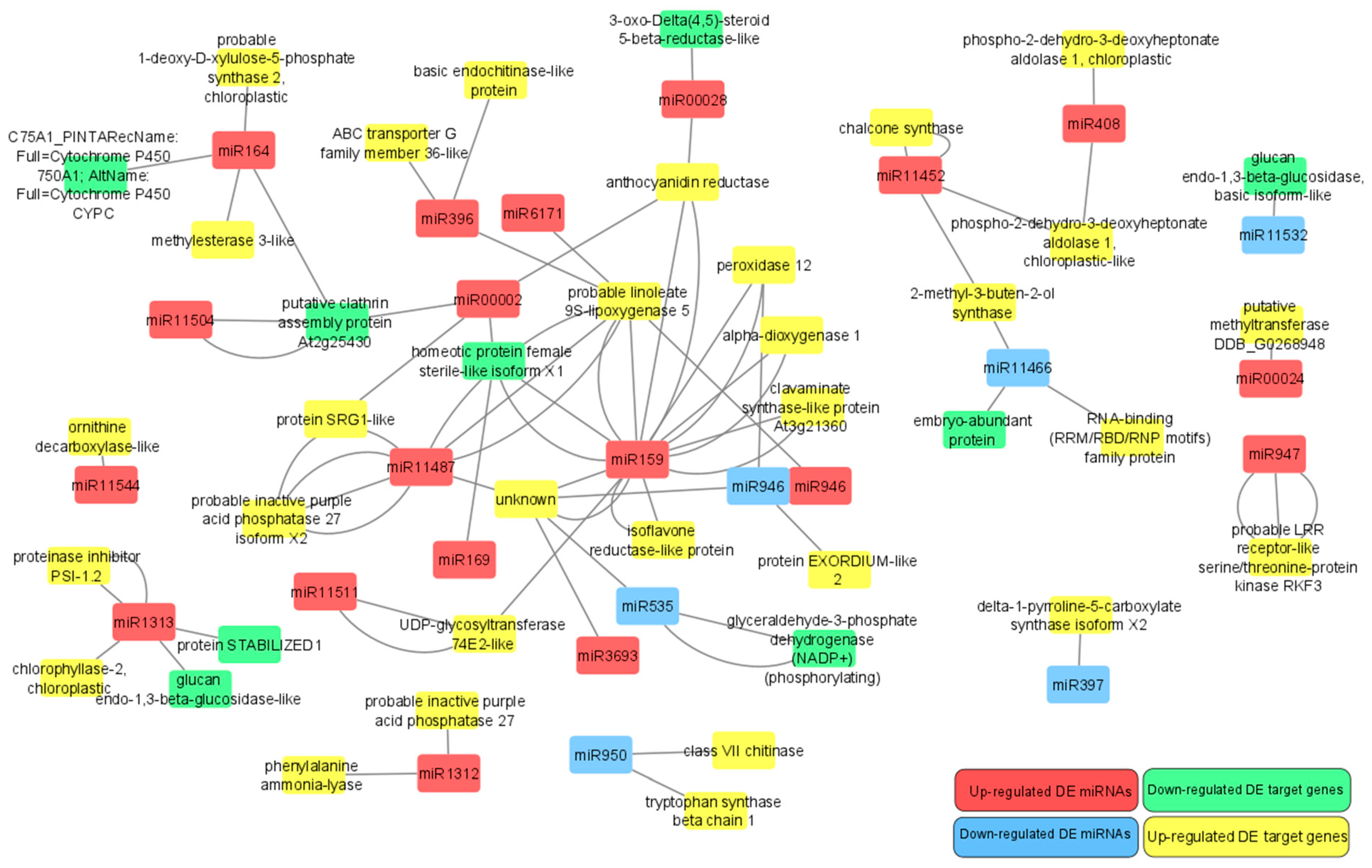
3.7. Analysis of Differentially Expressed microRNAs Targeting Differentially Expressed Gene Transcripts
4. Discussion
4.1. Differentially Expressed microRNAs in Conifers After Methyl Jasmonate Treatment
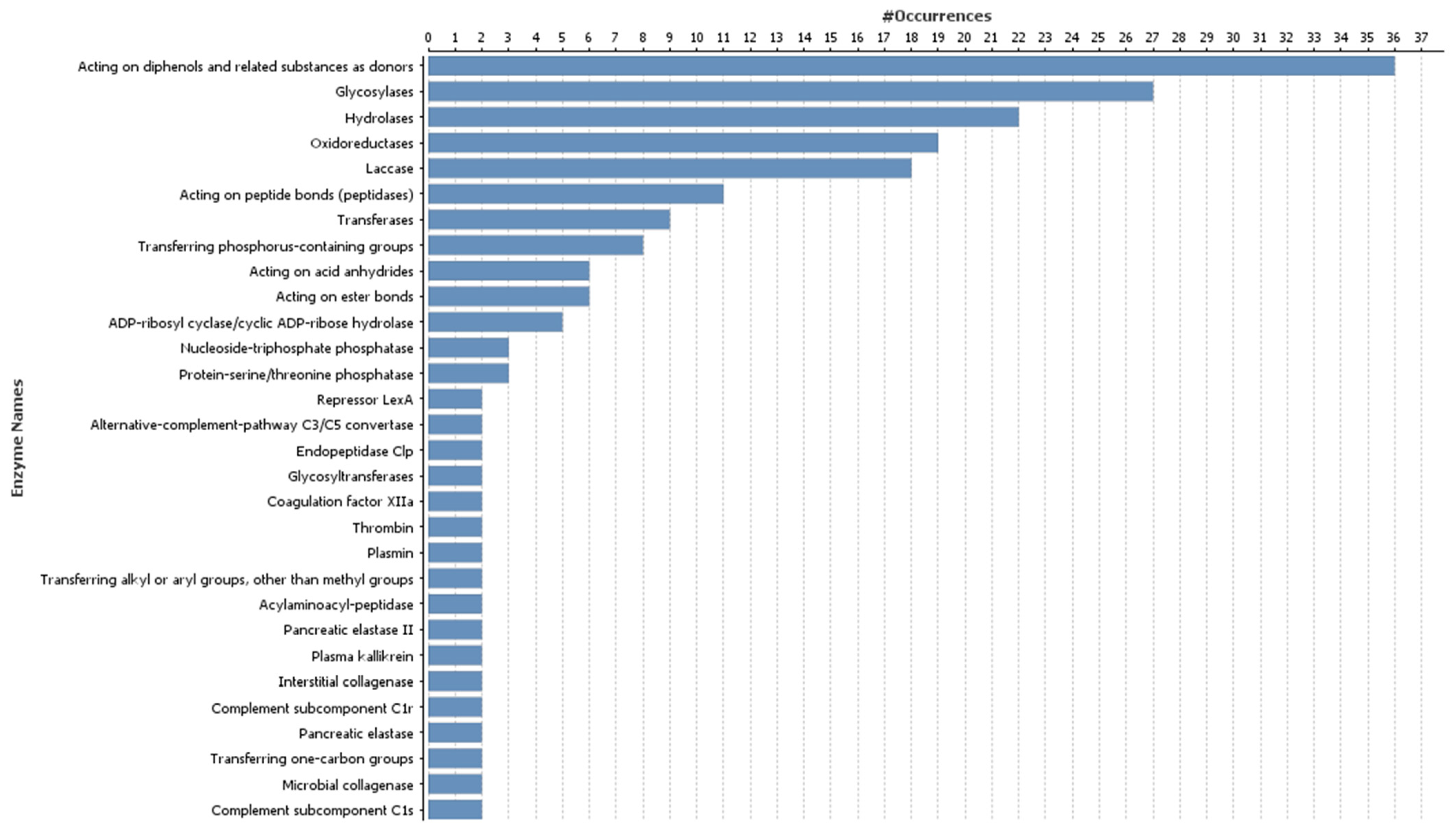
4.2. Predicted Target Genes for Differentially Expressed Down-Regulated microRNAs
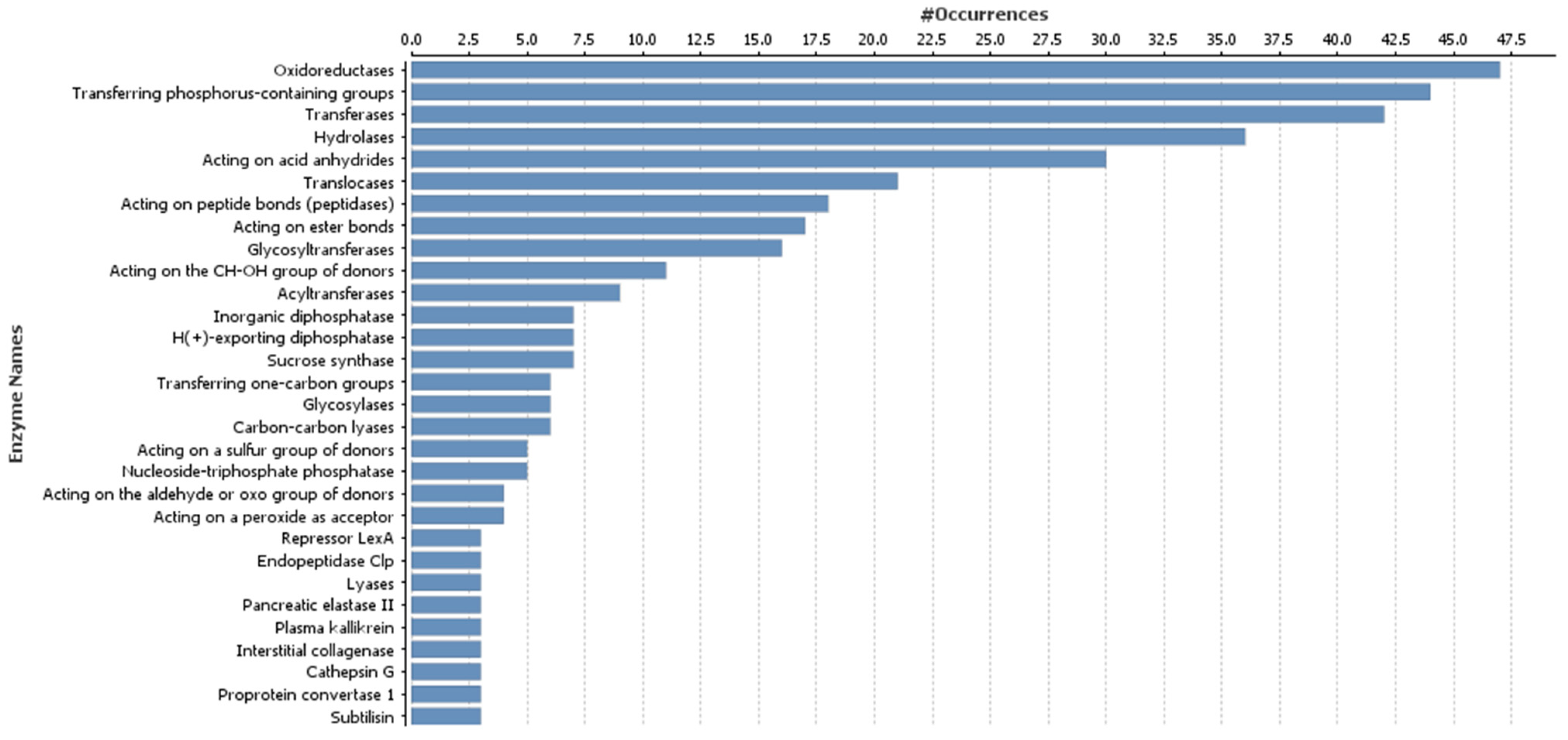
4.3. Predicted Target Genes for Differentially Expressed Up-Regulated microRNAs
4.4. Differentially Expressed Pinus sylvestris Genes Targeted by Differentially Expressed microRNAs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, T.; Pausas, J.G.; Belcher, C.M.; Schwilk, D.W.; Lamont, B.B. Fire-adapted traits of Pinus arose in the fiery Cretaceous. New Phytol. 2012, 194, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Rundel, P.W. A neogene heritage: Conifer distributions and endemism in mediterranean-climate ecosystems. Front. Ecol. Evol. 2019, 7, 364. [Google Scholar] [CrossRef]
- De La Torre, A.R.; Birol, I.; Bousquet, J.; Ingvarsson, P.K.; Jansson, S.; Jones, S.J.M.; Keeling, C.I.; MacKay, J.; Nilsson, O.; Ritland, K.; et al. Insights into conifer giga-genomes. Plant Physiol. 2014, 166, 1724–1732. [Google Scholar] [CrossRef] [PubMed]
- Houston Durrant, T.; de Rigo, D.; Caudullo, G. Pinus sylvestris. In European Atlas of Forest Tree Species; San-Miguel-Ayanz, J., de Rigo, D., Caudullo, G., Houston Durrant, T., Mauri, A., Eds.; Publication Office EU: Luxembourg, 2016; pp. 845–846. [Google Scholar]
- Lev-Yadun, S.; Sederoff, R. Pines as model gymnosperms to study evolution, wood formation, and perennial growth. J. Plant Growth Regul. 2000, 19, 290–305. [Google Scholar] [CrossRef]
- Fuchs, J.; Jovtchev, G.; Schubert, I. The chromosomal distribution of histone methylation marks in gymnosperms differs from that of angiosperms. Chromosom. Res. 2008, 16, 891–898. [Google Scholar] [CrossRef]
- Pyhäjärvi, T.; Kujala, S.T.; Savolainen, O. 275 years of forestry meets genomics in Pinus sylvestris. Evol. Appl. 2020, 13, 11–30. [Google Scholar] [CrossRef]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway spruce genome sequence and conifer genome evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef]
- Zimin, A.; Stevens, K.A.; Crepeau, M.W.; Holtz-Morris, A.; Koriabine, M.; Marçais, G.; Puiu, D.; Roberts, M.; Wegrzyn, J.L.; de Jong, P.J.; et al. Sequencing and assembly of the 22-Gb loblolly pine genome. Genetics 2014, 196, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Stevens, K.A.; Wegrzyn, J.L.; Zimin, A.; Puiu, D.; Crepeau, M.; Cardeno, C.; Paul, R.; Gonzalez-Ibeas, D.; Koriabine, M.; Holtz-Morris, A.E.; et al. Sequence of the sugar pine megagenome. Genetics 2016, 204, 1613–1626. [Google Scholar] [CrossRef] [PubMed]
- Voronova, A.; Rendón-Anaya, M.; Ingvarsson, P.; Kalendar, R.; Ruņģis, D. Comparative study of pine reference genomes reveals transposable element interconnected gene networks. Genes 2020, 11, 1216. [Google Scholar] [CrossRef]
- Carrington, J.C.; Ambros, V. Role of microRNAs in plant and animal development. Science 2003, 301, 336–338. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, R.; Li, N. MicroRNAs from plants to animals, do they define a new messenger for communication? Nutr. Metab. 2018, 15, 68. [Google Scholar] [CrossRef]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423–437. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol. 2006, 57, 19–53. [Google Scholar] [CrossRef]
- Brodersen, P.; Sakvarelidze-Achard, L.; Bruun-Rasmussen, M.; Dunoyer, P.; Yamamoto, Y.Y.; Sieburth, L.; Voinnet, O. Widespread translational inhibition by plant miRNAs and siRNAs. Science 2008, 320, 1185–1190. [Google Scholar] [CrossRef]
- Lanet, E.; Delannoy, E.; Sormani, R.; Floris, M.; Brodersen, P.; Crété, P.; Voinnet, O.; Robaglia, C. Biochemical Evidence for Translational Repression by arabidopsis MicroRNAs. Plant Cell 2009, 21, 1762–1768. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Staswick, P.E.; Su, W.; Howell, S.H. Methyl jasmonate inhibition of root growth and induction of a leaf protein are decreased in an Arabidopsis thaliana mutant. Proc. Natl. Acad. Sci. USA 1992, 89, 6837–6840. [Google Scholar] [CrossRef] [PubMed]
- Wasternack, C. Jasmonates: An update on biosynthesis, signal transduction and action in plant stress response, growth and development. Ann. Bot. 2007, 100, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.D.; Feys, B.F.; James, S.; Nieto-Rostro, M.; Turner, J. COI1: An Arabidopsis Gene Required for Jasmonate-Regulated Defense and Fertility. Science 1998, 280, 1091–1094. [Google Scholar] [CrossRef]
- Stintzi, A.; Browse, J. The Arabidopsis male-sterile mutant, opr3, lacks the 12-oxophytodienoic acid reductase required for jasmonate synthesis. Proc. Natl. Acad. Sci. USA 2000, 97, 10625–10630. [Google Scholar] [CrossRef]
- Ishiguro, S.; Kawai-Oda, A.; Ueda, J.; Nishida, I.; Okada, K. The defective in anther dehiscence1 gene encodes a novel phospholipase A1 catalyzing the initial step of jasmonic acid biosynthesis, which synchronizes pollen maturation, anther dehiscence, and flower opening in Arabidopsis. Plant Cell 2001, 13, 2191–2209. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Halitschke, R.; Kim, H.B.; Baldwin, I.T.; Feldmann, K.A.; Feyereisen, R. A knock-out mutation in allene oxide synthase results in male sterility and defective wound signal transduction in Arabidopsis due to a block in jasmonic acid biosynthesis. Plant J. 2002, 31, 1–12. [Google Scholar] [CrossRef]
- Xu, L.; Liu, F.; Lechner, E.; Genschik, P.; Crosby, W.L.; Ma, H.; Peng, W.; Huang, D.; Xie, D. The SCFCOI1 ubiquitin-ligase complexes are required for jasmonate response in Arabidopsis. Plant Cell 2002, 14, 1919–1935. [Google Scholar] [CrossRef] [PubMed]
- Thines, B.; Katsir, L.; Melotto, M.; Niu, Y.; Mandaokar, A.; Liu, G.; Nomura, K.; He, S.Y.; Howe, G.A.; Browse, J. JAZ repressor proteins are targets of the SCFCOI1 complex during jasmonate signalling. Nature 2007, 448, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, P.; Shockey, J.; Lévesque, C.A.; Cook, R.J.; Browse, J. A role for jasmonate in pathogen defense of Arabidopsis. Proc. Natl. Acad. Sci. USA 1998, 95, 7209–7214. [Google Scholar] [CrossRef]
- Chehab, E.W.; Yao, C.; Henderson, Z.; Kim, S.; Braam, J. Arabidopsis touch-induced morphogenesis is jasmonate mediated and protects against pests. Curr. Biol. 2012, 22, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Qiu, D.; Pan, X.; Wilson, I.W.; Li, F.; Liu, M.; Teng, W.; Zhang, B. High throughput sequencing technology reveals that the taxoid elicitor methyl jasmonate regulates microRNA expression in Chinese yew (Taxus chinensis). Gene 2009, 436, 37–44. [Google Scholar] [CrossRef]
- Wilkinson, S.W.; Vivian-Smith, A.; Krokene, P.; Mageroy, M.H. The microRNA response associated with methyl jasmonate-induced resistance in Norway spruce bark. Plant Gene 2021, 27, 100301. [Google Scholar] [CrossRef]
- Zhang, B.; Jin, Z.; Xie, D. Global Analysis of Non-coding Small RNAs in Arabidopsis in Response to Jasmonate Treatment by Deep Sequencing Technology. J. Integr. Plant Biol. 2012, 54, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Hoth, S.; Morgante, M.; Sanchez, J.P.; Hanafey, M.K.; Tingey, S.V.; Chua, N.H. Genome-wide gene expression profiling in Arabidopsis thaliana reveals new targets of abscisic acid and largerly impaired gene regulation in the abi1-1 mutant. J. Cell Sci. 2002, 115, 4891–4900. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, M.; Li, X.; Cao, B.; Ma, X. Identification of differentially expressed genes in leaf of Reaumuria soongorica under peg-induced drought stress by digital gene expression profiling. PLoS ONE 2014, 9, e94277, Correction in PLoS ONE 2014, 9, e104142. [Google Scholar] [CrossRef] [PubMed]
- Huynh, N.B.; Krokene, P.; Puentes, A.; Mageroy, M.H. Over 20 years of treating conifers with methyl jasmonate: Meta-analysis of effects on growth and resistance. For. Ecol. Manag. 2024, 561, 121893. [Google Scholar] [CrossRef]
- Mageroy, M.H.; Wilkinson, S.W.; Tengs, T.; Cross, H.; Almvik, M.; Pétriacq, P.; Vivian-Smith, A.; Zhao, T.; Fossdal, C.G.; Krokene, P. Molecular underpinnings of methyl jasmonate-induced resistance in Norway spruce. Plant. Cell Environ. 2020, 43, 1827–1843. [Google Scholar] [CrossRef]
- Šņepste, I.; Krivmane, B.; Šķipars, V.; Zaluma, A.; Ruņģis, D.E. Induction of Defense Responses in Pinus sylvestris Seedlings by Methyl Jasmonate and Response to Heterobasidion annosum and Lophodermium seditiosum Inoculation. Forests 2021, 12, 628. [Google Scholar] [CrossRef]
- Krivmane, B.; Šņepste, I.; Škipars, V.; Yakovlev, I.; Fossdal, C.G.; Vivian-Smith, A.; Ruņgis, D. Identification and in Silico Characterization of Novel and Conserved MicroRNAs in Methyl Jasmonate-Stimulated Scots Pine (Pinus sylvestris L.) Needles. Forests 2020, 11, 384. [Google Scholar] [CrossRef]
- Rubio-Piña, J.A.; Zapata-Pérez, O. Isolation of total RNA from tissues rich in polyphenols and polysaccharides of mangrove plants. Electron. J. Biotechnol. 2011, 14, 11. [Google Scholar] [CrossRef]
- Kānberga-Siliņa, K.; Rauda, E.; Šķipars, V.; Vivian-smith, A.; Yakovlev, I.; Krivmane, B.; Šņepste, I.; Ruņģis, D. Transcriptomic response to methyl jasmonate treatment of Scots pine (Pinus sylvestris) seedlings. Environ. Exp. Biol. 2017, 15, 257–274. [Google Scholar] [CrossRef]
- Wachowiak, W.; Trivedi, U.; Perry, A.; Cavers, S. Comparative transcriptomics of a complex of four European pine species. BMC Genom. 2015, 16, 234. [Google Scholar] [CrossRef]
- Šķipars, V.; Ruņģis, D. Transcript dynamics in wounded and inoculated Scots pine. Int. J. Mol. Sci. 2021, 22, 1505. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhao, P.X. PsRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Aksay, G.; Dolgosheina, E.; Ebhardt, H.A.; Magrini, V.; Mardis, E.R.; Sahinalp, S.C.; Unrau, P.J. Comparative analysis of the small RNA transcriptomes of Pinus contorta and Oryza sativa. Genome Res. 2008, 18, 571–584. [Google Scholar] [CrossRef]
- Lu, S.; Sun, Y.H.; Amerson, H.; Chiang, V.L. MicroRNAs in loblolly pine (Pinus taeda L.) and their association with fusiform rust gall development. Plant J. 2007, 51, 1077–1098. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.C.; Zhang, H.; Lu, S.; Zhang, L.; Qiu, Z.; Zhao, Y.; Zeng, Q.Y.; Lin, J. Transcriptome-wide identification and characterization of miRNAs from Pinus densata. BMC Genom. 2012, 13, 132. [Google Scholar] [CrossRef] [PubMed]
- Yakovlev, I.A.; Fossdal, C.G.; Johnsen, Ø. MicroRNAs, the epigenetic memory and climatic adaptation in Norway spruce. New Phytol. 2010, 187, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Krivmane, B.; Ruņģe, K.S.; Samsone, I.; Ruņģis, D.E. Differentially Expressed Conserved Plant Vegetative Phase-Change-Related microRNAs in Mature and Rejuvenated Silver Birch In Vitro Propagated Tissues. Plants 2023, 12, 1993. [Google Scholar] [CrossRef] [PubMed]
- Chávez Montes, R.A.; De Fátima Rosas-Cárdenas, F.; De Paoli, E.; Accerbi, M.; Rymarquis, L.A.; Mahalingam, G.; Marsch-Martínez, N.; Meyers, B.C.; Green, P.J.; De Folter, S. Sample sequencing of vascular plants demonstrates widespread conservation and divergence of microRNAs. Nat. Commun. 2014, 5, 3722. [Google Scholar] [CrossRef] [PubMed]
- Puentes, A.; Zhao, T.; Lundborg, L.; Björklund, N.; Borg-Karlson, A.K. Variation in Methyl Jasmonate-Induced Defense Among Norway Spruce Clones and Trade-Offs in Resistance Against a Fungal and an Insect Pest. Front. Plant Sci. 2021, 12, 678959. [Google Scholar] [CrossRef] [PubMed]
- Cuperus, J.T.; Fahlgren, N.; Carrington, J.C. Evolution and functional diversification of MIRNA genes. Plant Cell 2011, 23, 431–442. [Google Scholar] [CrossRef]
- Galdino, J.H.; Eguiluz, M.; Guzman, F.; Margis, R. Novel and conserved miRNAs among Brazilian pine and other gymnosperms. Front. Genet. 2019, 10, 222. [Google Scholar] [CrossRef]
- Sun, G.; Yang, Y.; Xie, F.; Wen, J.F.; Wu, J.; Wilson, I.W.; Tang, Q.; Liu, H.; Qiu, D. Deep Sequencing Reveals Transcriptome Re-Programming of Taxus × media Cells to the Elicitation with Methyl Jasmonate. PLoS ONE 2013, 8, e62865. [Google Scholar] [CrossRef]
- Bai, Y.; Ali, S.; Liu, S.; Zhou, J.; Tang, Y. Characterization of plant laccase genes and their functions. Gene 2023, 852, 147060. [Google Scholar] [CrossRef]
- Li, C.; Li, D.; Zhou, H.; Li, J.; Lu, S. Analysis of the laccase gene family and miR397-/miR408-mediated posttranscriptional regulation in Salvia miltiorrhiza. PeerJ 2019, 2019, e7605. [Google Scholar] [CrossRef]
- Elfstrand, M.; Baison, J.; Lundén, K.; Zhou, L.; Vos, I.; Capador, H.D.; Åslund, M.S.; Chen, Z.; Chaudhary, R.; Olson, Å.; et al. Association genetics identifies a specifically regulated Norway spruce laccase gene, PaLAC5, linked to Heterobasidion parviporum resistance. Plant Cell Environ. 2020, 43, 1779–1791. [Google Scholar] [CrossRef]
- Van Ghelder, C.; Parent, G.J.; Rigault, P.; Prunier, J.; Giguère, I.; Caron, S.; Stival Sena, J.; Deslauriers, A.; Bousquet, J.; Esmenjaud, D.; et al. The large repertoire of conifer NLR resistance genes includes drought responsive and highly diversified RNLs. Sci. Rep. 2019, 9, 11614. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Xu, J.; Arikit, S.; Meyers, B.C. Extensive families of miRNAs and PHAS loci in Norway spruce demonstrate the origins of complex phasiRNA networks in seed plants. Mol. Biol. Evol. 2015, 32, 2905–2918. [Google Scholar] [CrossRef]
- Zhang, S.; Yan, S.; Zhao, J.; Xiong, H.; An, P.; Wang, J.; Zhang, H.; Zhang, L. Identification of miRNAs and their target genes in Larix olgensis and verified of differential expression miRNAs. BMC Plant Biol. 2019, 19, 247. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.L.; Yuan, S.N.; Zhang, H.N.; Zhang, Y.Y.; Zhang, Y.J.; Wang, G.Y.; Li, Y.Q.; Li, G.L. Heat-response patterns of the heat shock transcription factor family in advanced development stages of wheat (Triticum aestivum L.) and thermotolerance-regulation by TaHsfA2-10. BMC Plant Biol. 2020, 20, 364. [Google Scholar] [CrossRef] [PubMed]
- Kunieda, T.; Fujiwara, T.; Amano, T.; Shioi, Y. Molecular cloning and characterization of a senescence-induced tau-class glutathione S-transferase from barley leaves. Plant Cell Physiol. 2005, 46, 1540–1548. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, L.; Zhao, W.; Fu, L.; Han, Y.; Wang, K.; Yan, L.; Li, Y.; Zhang, X.H.; Min, D.H. Genome-wide analysis of the serine carboxypeptidase-like protein family in Triticum aestivum reveals TaSCPL184-6D is involved in abiotic stress response. BMC Genom. 2021, 22, 350. [Google Scholar] [CrossRef] [PubMed]
- Ciftci-Yilmaz, S.; Mittler, R. The zinc finger network of plants. Cell Mol. Life Sci. 2008, 65, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Larsen, B.; Xu, D.; Halkier, B.A.; Nour-Eldin, H.H. Advances in methods for identification and characterization of plant transporter function. J. Exp. Bot. 2017, 68, 4045–4056. [Google Scholar] [CrossRef] [PubMed]
- Romero-Hernandez, G.; Martinez, M. Plant Kinases in the Perception and Signaling Networks Associated with Arthropod Herbivory. Front. Plant Sci. 2022, 13, 824422. [Google Scholar] [CrossRef] [PubMed]
- Goff, K.E.; Ramonell, K.M. The Role and Regulation of Receptor-Like Kinases in Plant Defense. Gene Regul. Syst. Biol. 2007, 1, 117762500700100. [Google Scholar] [CrossRef]
- Lee, H.J.; Park, O.K. Lipases associated with plant defense against pathogens. Plant Sci. 2019, 279, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.R.; Iriyama, R.; Fernando, D.D. Expression patterns of conserved microRNAs in the male gametophyte of loblolly pine (Pinus taeda). Plant Reprod. 2014, 27, 69–78. [Google Scholar] [CrossRef]
- Kartashov, A.V.; Zlobin, I.E.; Pashkovskiy, P.P.; Pojidaeva, E.S.; Ivanov, Y.V.; Mamaeva, A.S.; Fesenko, I.A.; Kuznetsov, V.V. Quantitative analysis of differential dehydrin regulation in pine and spruce seedlings under water deficit. Plant Physiol. Biochem. 2021, 162, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Li, S.; Shi, A.; Li, Y.; He, C.; Yan, Y.; Wang, J.; Sun, M.; Yu, X. Genome-wide analysis of the AINTEGUMENTA-like (AIL) transcription factor gene family in pumpkin (Cucurbita moschata Duch.) and CmoANT1.2 response in graft union healing. Plant Physiol. Biochem. 2021, 162, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.U.; Hayward, A.; Irihimovitch, V.; Fletcher, S.; Tanurdzic, M.; Pocock, A.; Beveridge, C.A.; Mitter, N. Juvenility and vegetative phase transition in tropical/subtropical tree crops. Front. Plant Sci. 2019, 10, 729. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-H.; Wu, T.; Han, S.-Y.; Qi, L.; Wang, Z.S. Expression Analysis of 12 miRNA Families Specific to Conifers during Somatic Embryogenesis of Larch. For. Res. 2012, 25, 411–418. [Google Scholar]
- Rodrigues, A.S.; Chaves, I.; Costa, B.V.; Lin, Y.C.; Lopes, S.; Milhinhos, A.; Van de Peer, Y.; Miguel, C.M. Small RNA profiling in Pinus pinaster reveals the transcriptome of developing seeds and highlights differences between zygotic and somatic embryos. Sci. Rep. 2019, 9, 11327. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ng, T.B. Isolation of an antifungal thaumatin-like protein from kiwi fruits. Phytochemistry 2002, 61, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Henarejos, S.A.; Alcaraz, L.A.; Donaire, A. Blue Copper Proteins: A rigid machine for efficient electron transfer, a flexible device for metal uptake. Arch. Biochem. Biophys. 2015, 584, 134–148. [Google Scholar] [CrossRef]
- De Rienzo, F.; Gabdoulline, R.R.; Menziani, M.C.; Wade, R.C. Blue copper proteins: A comparative analysis of their molecular interaction properties. Protein Sci. 2000, 9, 1439–1454. [Google Scholar] [CrossRef] [PubMed]
- Valdés-López, O.; Yang, S.S.; Aparicio-Fabre, R.; Graham, P.H.; Reyes, J.L.; Vance, C.P.; Hernández, G. MicroRNA expression profile in common bean (Phaseolus vulgaris) under nutrient deficiency stresses and manganese toxicity. New Phytol. 2010, 187, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Stein, O.; Granot, D. An overview of sucrose synthases in plants. Front. Plant Sci. 2019, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Niu, S.; El-Kassaby, Y.A.; Li, W. Genome-wide identification of late embryogenesis abundant protein family and their key regulatory network in Pinus tabuliformis cold acclimation. Tree Physiol. 2023, 43, 1964–1985. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.A.; Sabeem, M.; Mullath, S.K.; Brini, F.; Masmoudi, K. Plant group ii lea proteins: Intrinsically disordered structure for multiple functions in response to environmental stresses. Biomolecules 2021, 11, 1662. [Google Scholar] [CrossRef]
- Graether, S.P. Proteins Involved in Plant Dehydration Protection: The Late Embryogenesis Abundant Family. Biomolecules 2022, 12, 10–12. [Google Scholar] [CrossRef]
- Lehti-Shiu, M.D.; Panchy, N.; Wang, P.; Uygun, S.; Shiu, S.H. Diversity, expansion, and evolutionary novelty of plant DNA-binding transcription factor families. Biochim. Biophys. Acta—Gene Regul. Mech. 2017, 1860, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Blazek, E.; Mittler, G.; Meisterernst, M. The mediator of RNA polymerase II. Chromosoma 2005, 113, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Li, K.; Li, Y.; Zhao, X.; Wang, L. MYB transcription factors as regulators of secondary metabolism in plants. Biology 2020, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Xu, Y.; Shi, M.; Lai, Y.; Wu, X.; Wang, H.; Zhu, Z.; Scott Poethig, R.; Wu, G. Repression of miR156 by miR159 regulates the timing of the juvenile-to-adult transition in Arabidopsis. Plant Cell 2017, 29, 1293–1304. [Google Scholar] [CrossRef]
- Hu, H.; Guo, Z.; Yang, J.; Cui, J.; Zhang, Y.; Xu, J. Transcriptome and microRNA Sequencing Identified miRNAs and Target Genes in Different Developmental Stages of the Vascular Cambium in Cryptomeria fortunei Hooibrenk. Front. Plant Sci. 2021, 12, 751771. [Google Scholar] [CrossRef] [PubMed]
- Afzal, A.J.; Wood, A.J.; Lightfoot, D.A. Plant receptor-like serine threonine kinases: Roles in signaling and plant defense. Mol. Plant-Microbe Interact. 2008, 21, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Leubner-Metzger, G. Functions and regulation of β-1,3-glucanases during seed germination, dormancy release and after-ripening. Seed Sci. Res. 2003, 13, 17–34. [Google Scholar] [CrossRef]
- Liu, B.; Xue, X.; Cui, S.; Zhang, X.; Han, Q.; Zhu, L.; Liang, X.; Wang, X.; Huang, L.; Chen, X.; et al. Cloning and characterization of a wheat b-1,3-glucanase gene induced by the stripe rust pathogen Puccinia striiformis f. sp. tritici. Mol. Biol. Rep. 2010, 37, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, X.Y.; Guo, W.Z. The cytochrome P450 superfamily: Key players in plant development and defense. J. Integr. Agric. 2015, 14, 1673–1686. [Google Scholar] [CrossRef]
- Pandian, B.A.; Sathishraj, R.; Djanaguiraman, M.; Prasad, P.V.V.; Jugulam, M. Role of cytochrome P450 enzymes in plant stress response. Antioxidants 2020, 9, 454. [Google Scholar] [CrossRef] [PubMed]
- Heitz, T.; Widemann, E.; Lugan, R.; Miesch, L.; Ullmann, P.; Désaubry, L.; Holder, E.; Grausem, B.; Kandel, S.; Miesch, M.; et al. Cytochromes P450 CYP94C1 and CYP94B3 catalyze two successive oxidation steps of plant hormone jasmonoyl-isoleucine for catabolic turnover. J. Biol. Chem. 2012, 287, 6296–6306. [Google Scholar] [CrossRef]
- Koo, A.J.K.; Howe, G.A. Catabolism and deactivation of the lipid-derived hormone jasmonoyl-isoleucine. Front. Plant Sci. 2012, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Hamberger, B.; Ohnishi, T.; Hamberger, B.; Séguin, A.; Bohlmann, J. Evolution of diterpene metabolism: Sitka spruce CYP720B4 catalyzes multiple oxidations in resin acid biosynthesis of conifer defense against insects. Plant Physiol. 2011, 157, 1677–1695. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| microRNA Family | microRNA Sequence | Mature microRNA Length (in Nucleotides) | Count of Precursor-microRNAs | Precursor-microRNA Accession Number |
|---|---|---|---|---|
| Up-regulated | ||||
| miR00002 | GTTCGGAGAAATGCGTGCTCG | 21 | 1 | TC188772 |
| miR00024 | CGTGTTCCCAGGTCGCCCGG | 20 | 1 | TC188772 |
| miR00024 | CGTGTTCCCAGGTCACCCAG | 20 | 1 | TC188772 |
| miR00024 | CGTGTTCCCAGGTCGCCCCGG | 21 | 1 | TC188772 |
| miR00028 | ATGTGAGAAAAGGGTTTGTGG | 21 | 1 | AW010185 |
| miR11452 * | TGGGAGCGATCGATGAGGTGTT | 22 | - | - |
| miR11466 * | CGATCTCCAGAAGACACTTGT | 21 | - | - |
| miR11487 * | CATATTGATCGCCTTCTCAGT | 21 | - | - |
| miR11487 * | TCATATTGATCGCCTTCTCAG | 21 | - | - |
| miR11487 * | TGAGAAAGCGATCAACATGAC | 21 | - | - |
| miR11487 * | TGAGAAGGCGATCAACATGAC | 21 | - | - |
| miR11487 * | TGAGAAGGCGATCAACATGAT | 21 | - | - |
| miR11487 * | TGAGAAGGCGATCAACATGC | 20 | - | - |
| miR11504 * | AATGAGCTCCTCCTCATGTCC | 21 | - | - |
| miR11504 * | AATGAGCTCCTCCTCATGTCT | 21 | - | - |
| miR11511 * | TCCAACGAAGATCAGAAAGG | 21 | - | - |
| miR11511 * | TCCAACGAAGATCAGAAGGC | 20 | - | - |
| miR11511 * | TCCAACGAAGATCAGAAGGCGT | 22 | - | - |
| miR11511 * | TCCAACGAAGATCAGAAGGCTC | 22 | - | - |
| miR11511 * | TCCACCGAAGATCAGAAGGTTC | 22 | - | - |
| miR11511 * | TCCACCGAAGATCAGAAGGTTTT | 23 | - | - |
| miR11544 * | TGGAGCTGTTGTCACTCCACT | 21 | - | - |
| miR11551 * | TGTTTTGCTTTCCCTCCGCAAT | 22 | - | - |
| miR1312 * | TCGGAGAGAATATGGCGAGAT | 21 | - | - |
| miR1313 * | AACAATAATTTCAGTGGAAGA | 21 | 2 | CO167297, DR013032 |
| miR1314 * | TCGGCCTCGAAATGTTAGGAGAA | 23 | - | - |
| miR159 | TTTGGTTTGAAGGGAGCTCCA | 21 | - | - |
| miR159 | CTTGGATTGAAGGGAGCTCC | 20 | 1 | DR078315 |
| miR159 | TTTGGTTTGAAGGGAGCTCT | 20 | - | - |
| miR164 | TGGAGAAGCAGGGCACGTGCG | 21 | - | - |
| miR169 | GGCAAGTTGTTCTTGGCAAAG | 21 | - | - |
| miR3693 * | CTGAACTGCTTATAGATGGGA | 21 | - | - |
| miR3693 * | TGAACTGCTTATAGATGGGAG | 21 | - | - |
| miR396 | CTCAAAGAAAGCTGTGGGAAA | 21 | - | - |
| miR398 | ACGTGTTCCCAGGTCACCCCA | 21 | 2 | TC184482, TC188772 |
| miR398 | CGTGTTCCCAGGTCACCCCAG | 21 | 2 | TC184482, TC188772 |
| miR408 | TGCACTGCCTCTTCCCTGGCT | 21 | 2 | TC159922, TC180357 |
| miR6171 | TGTGGATTGCTGAAGGATTTA | 21 | - | - |
| miR946 * | TGTGGATAGAGAAGGGTTAGT | 21 | - | - |
| miR947 * | CATCGGAATCTGTTACTGTTTT | 22 | - | - |
| miR947 * | CATCGGAATCTGTTACTGTTTC | 22 | - | - |
| miR947 * | CATCGGAATCTGTTACTGTTT | 21 | - | - |
| miR950 * | TCTGGTCCTCGGTGGTTTATGAAT | 24 | - | - |
| Down-regulated | ||||
| miR11466 * | CACTTCCAATAGACACTTGTT | 21 | - | - |
| miR11466 * | TCTACTTCCACAAGACACTTGCC | 23 | - | - |
| miR11490 * | TAGCCCGAACGCACAATTGGA | 21 | - | - |
| miR11532 * | TGACATTGTAAAGTACGGGAAT | 22 | - | - |
| miR3701 * | TAAACAATGTCCACCCTTCATT | 22 | - | - |
| miR397 | TCATCATTGAGTGCAGCATTG | 21 | - | - |
| miR535 | TCGACAACGAGAGAGAGCACGC | 22 | - | - |
| miR946 * | TCAGACCTTCTCCTATCCACAAT | 23 | - | - |
| miR946 * | CAGCCCTTCTCCTATCCACCAAC | 23 | - | - |
| miR946 * | TAGACCTTCTCCTATCCACCAAT | 23 | - | - |
| miR950 * | TCACATCTAGGCCACGATGGTT | 22 | - | - |
| miR950 * | TCACGTCTGGGCCTCTATGGTT | 22 | - | - |
| miR950 * | TCGTGTCCTCGGTGGTTTATGA | 22 | - | - |
| miR950 * | TTTACGTCTGGTCCTCGATGGTT | 23 | - | - |
| miR950 * | TTACATCTGGGCCACGGTGGTT | 22 | 1 | TC178188 |
| microRNA Family | Pinus sylvestris | Taxus chinensis | Taxus × Media | Picea abies |
|---|---|---|---|---|
| miR156 | - | down | down | - |
| miR159 | up | - | - | - |
| miR162 | - | - | - | down |
| miR164 | up | up | up | - |
| miR167 | - | - | - | down |
| miR168 | - | down | down | - |
| miR169 | up | down | down | up |
| miR172 | - | down | down | up |
| miR319 | - | - | - | down |
| miR390 | - | up | up | up |
| miR396 | up | down | down | down |
| miR397 | down | - | - | up |
| miR398 | up | - | - | up |
| miR398 | - | - | - | up |
| miR399 | - | - | - | - |
| miR408 | up | - | down | - |
| miR437 | - | - | - | up |
| miR480 | - | down | - | - |
| miR482 | - | - | - | up |
| miR529 | - | - | - | down |
| miR535 | down | - | - | - |
| miR858 | - | - | - | up |
| miR946 | down/up | - | - | - |
| miR947 | up | - | - | - |
| miR950 | down/up | - | - | up |
| miR1310 | - | down | - | - |
| miR1311 | - | - | - | up |
| miR1312 | up | - | - | - |
| miR1313 | up | - | - | down |
| miR1314 | up | - | - | - |
| miR3693 | up | - | - | up |
| miR3701 | down | - | - | up |
| miR4376 | - | - | - | up |
| miR6171 | up | - | - | - |
| miR00002 | up | - | - | - |
| miR00024 | up | - | - | - |
| miR00028 | up | - | - | - |
| miR11452 | up | - | - | - |
| miR11466 | down/up | - | - | - |
| miR11467 | - | - | - | down |
| miR11487 | up | - | - | up |
| miR11490 | down | - | - | - |
| miR11498 | - | - | - | down |
| miR11504 | up | - | - | - |
| miR11511 | up | - | - | - |
| miR11524 | - | - | - | up |
| miR11532 | down | - | - | - |
| miR11539 | - | - | - | up |
| miR11544 | up | - | - | - |
| miR11551 | up | - | - | up |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krivmane, B.; Ruņģis, D.E. Differential microRNA and Target Gene Expression in Scots Pine (Pinus sylvestris L.) Needles in Response to Methyl Jasmonate Treatment. Genes 2025, 16, 26. https://doi.org/10.3390/genes16010026
Krivmane B, Ruņģis DE. Differential microRNA and Target Gene Expression in Scots Pine (Pinus sylvestris L.) Needles in Response to Methyl Jasmonate Treatment. Genes. 2025; 16(1):26. https://doi.org/10.3390/genes16010026
Chicago/Turabian StyleKrivmane, Baiba, and Dainis Edgars Ruņģis. 2025. "Differential microRNA and Target Gene Expression in Scots Pine (Pinus sylvestris L.) Needles in Response to Methyl Jasmonate Treatment" Genes 16, no. 1: 26. https://doi.org/10.3390/genes16010026
APA StyleKrivmane, B., & Ruņģis, D. E. (2025). Differential microRNA and Target Gene Expression in Scots Pine (Pinus sylvestris L.) Needles in Response to Methyl Jasmonate Treatment. Genes, 16(1), 26. https://doi.org/10.3390/genes16010026