
Novel Splice-Altering Variants in the CHM and CACNA1F Genes Causative of X-Linked Choroideremia and Cone Dystrophy

 ,
,  , , ,
, , ,  , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
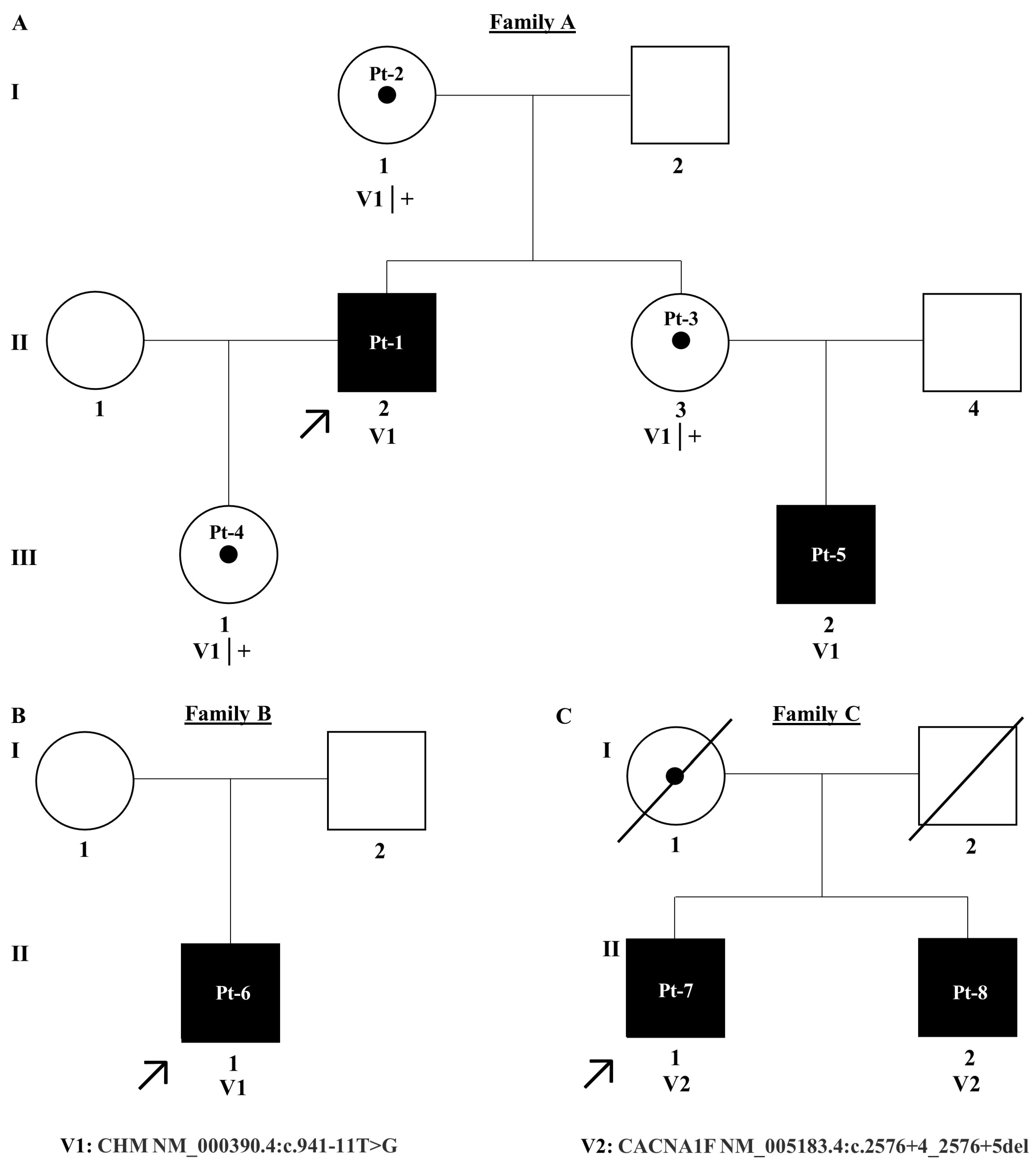
2.1. Patient Recruitment
2.2. DNA Isolation and Target 5000 Next-Generation Sequencing
2.3. Cascade Analysis
2.4. Variant Interpretation
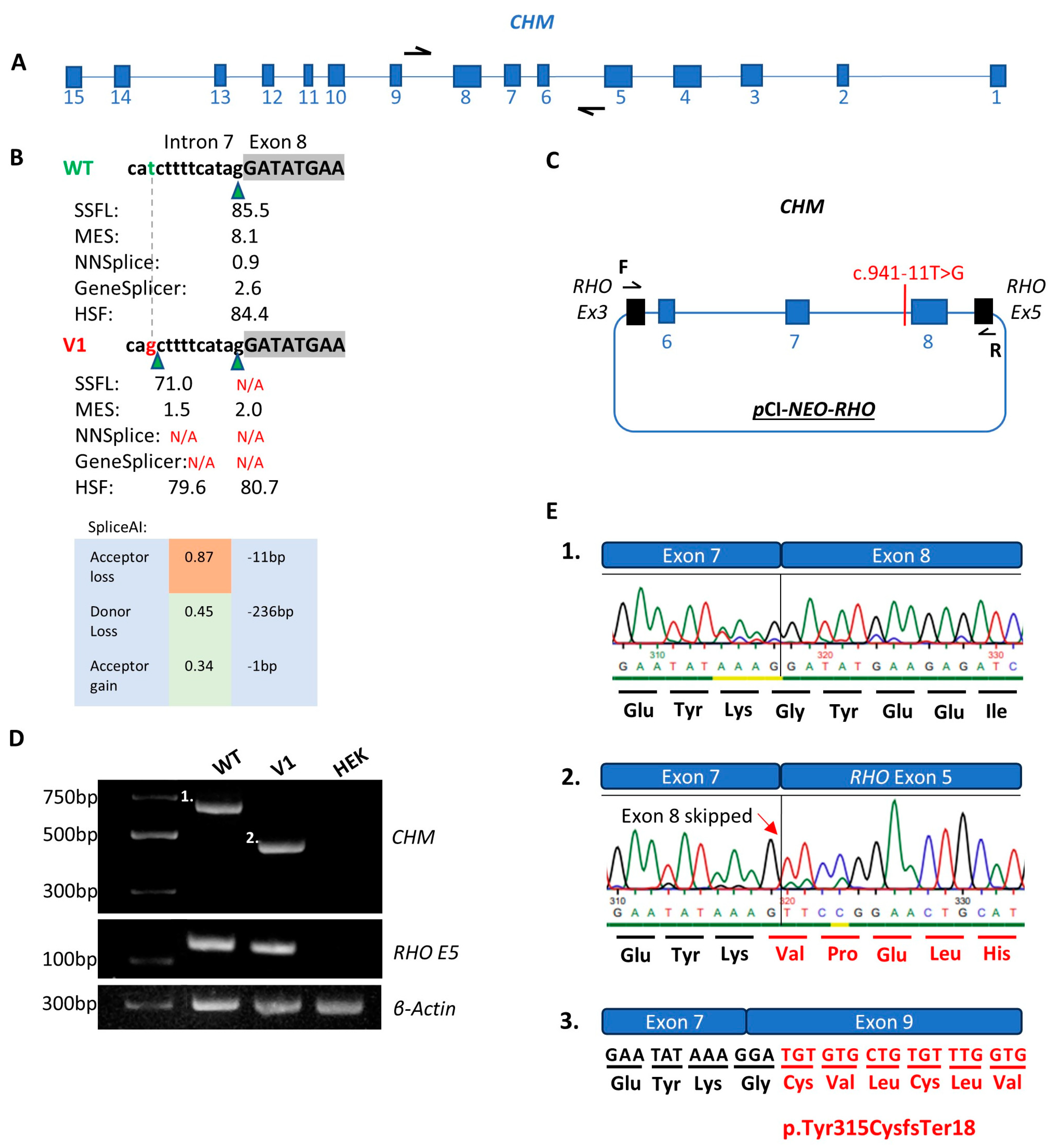
2.5. Midigene Generation
2.6. Midigene Expression Analysis
2.7. RT-PCR
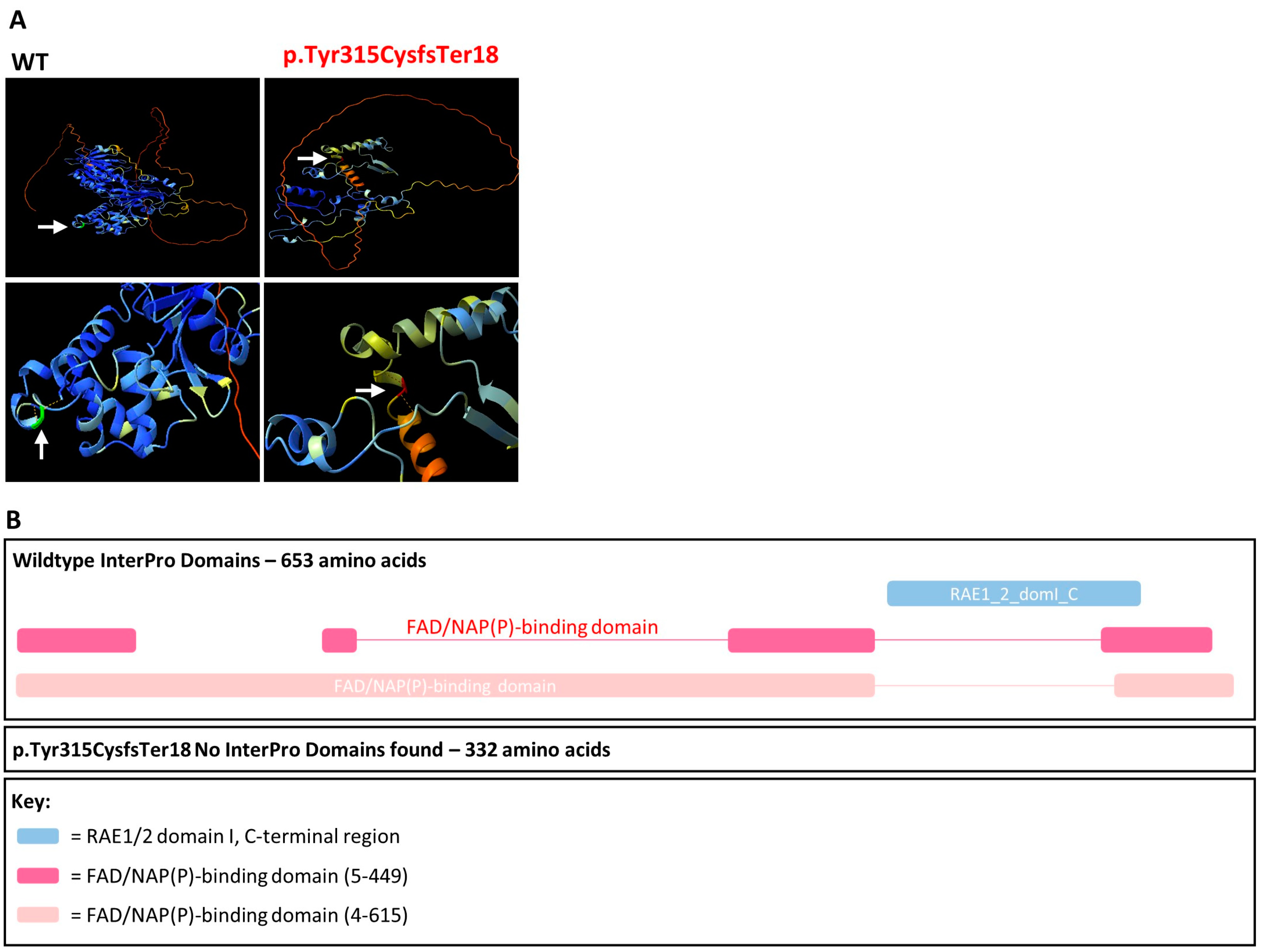
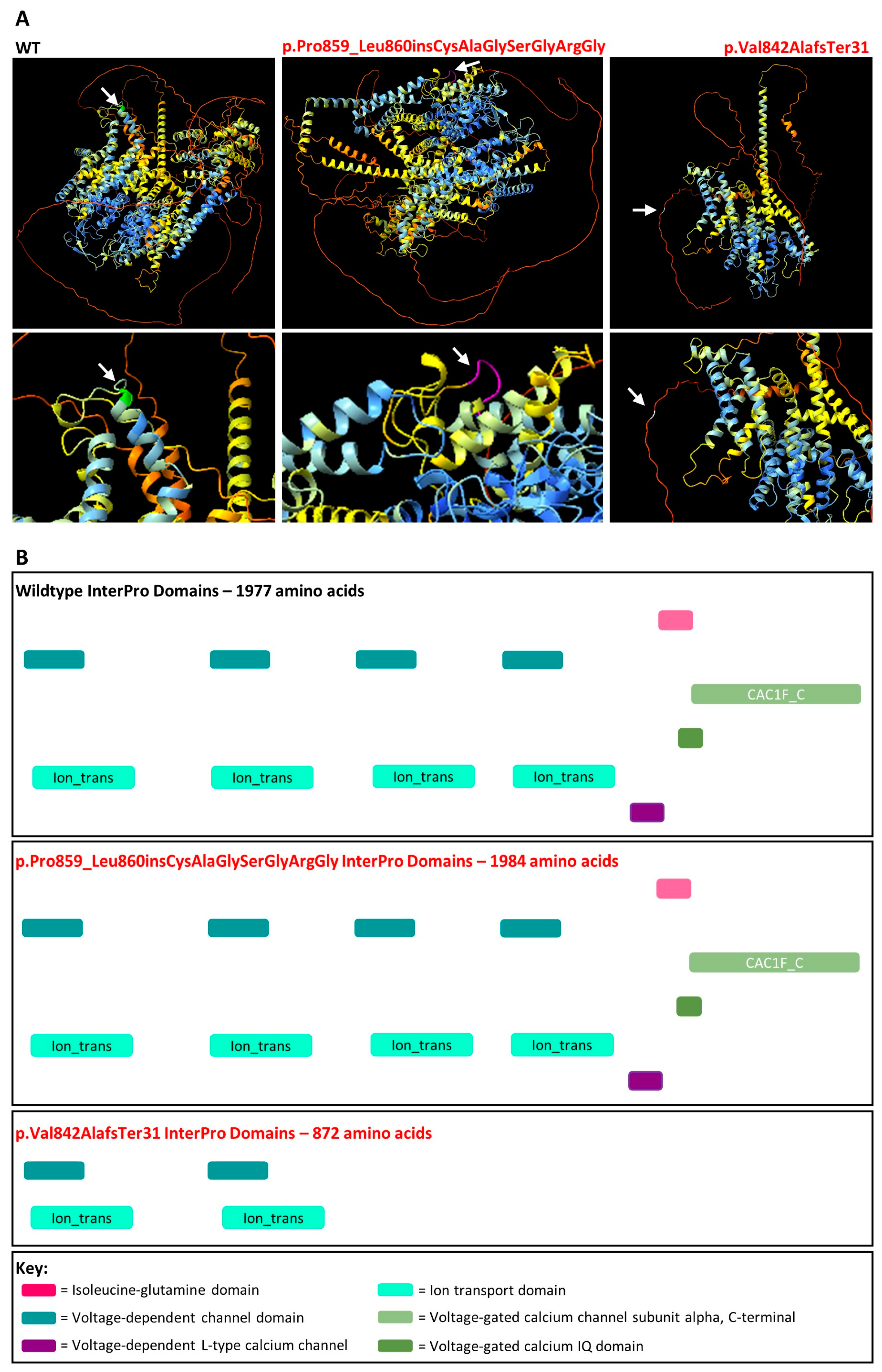
2.8. AlphaFold Protein Modelling and InterPro Domains
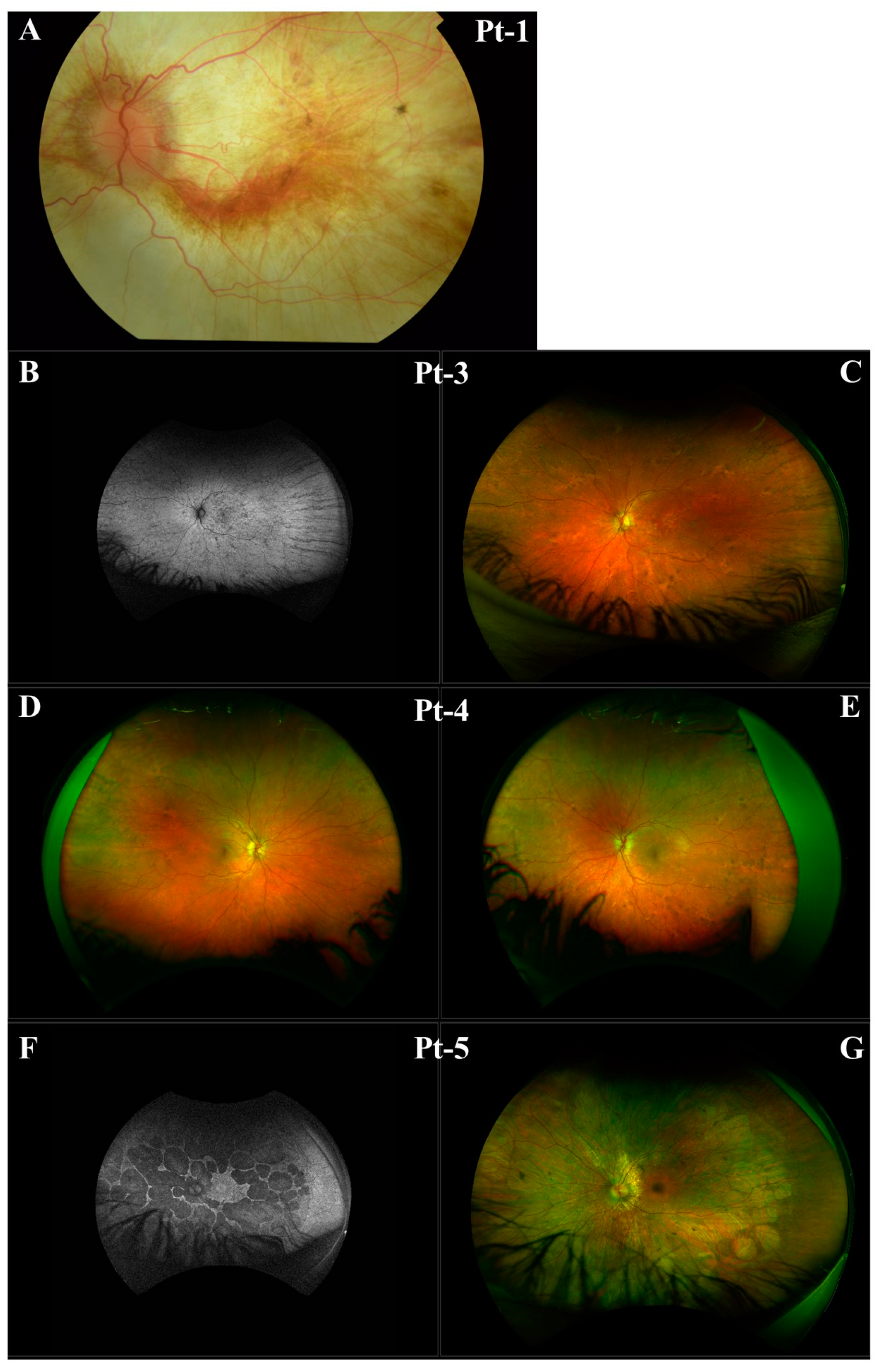
3. Results
3.1. CHM NM_000390.4:c.941-11T>G
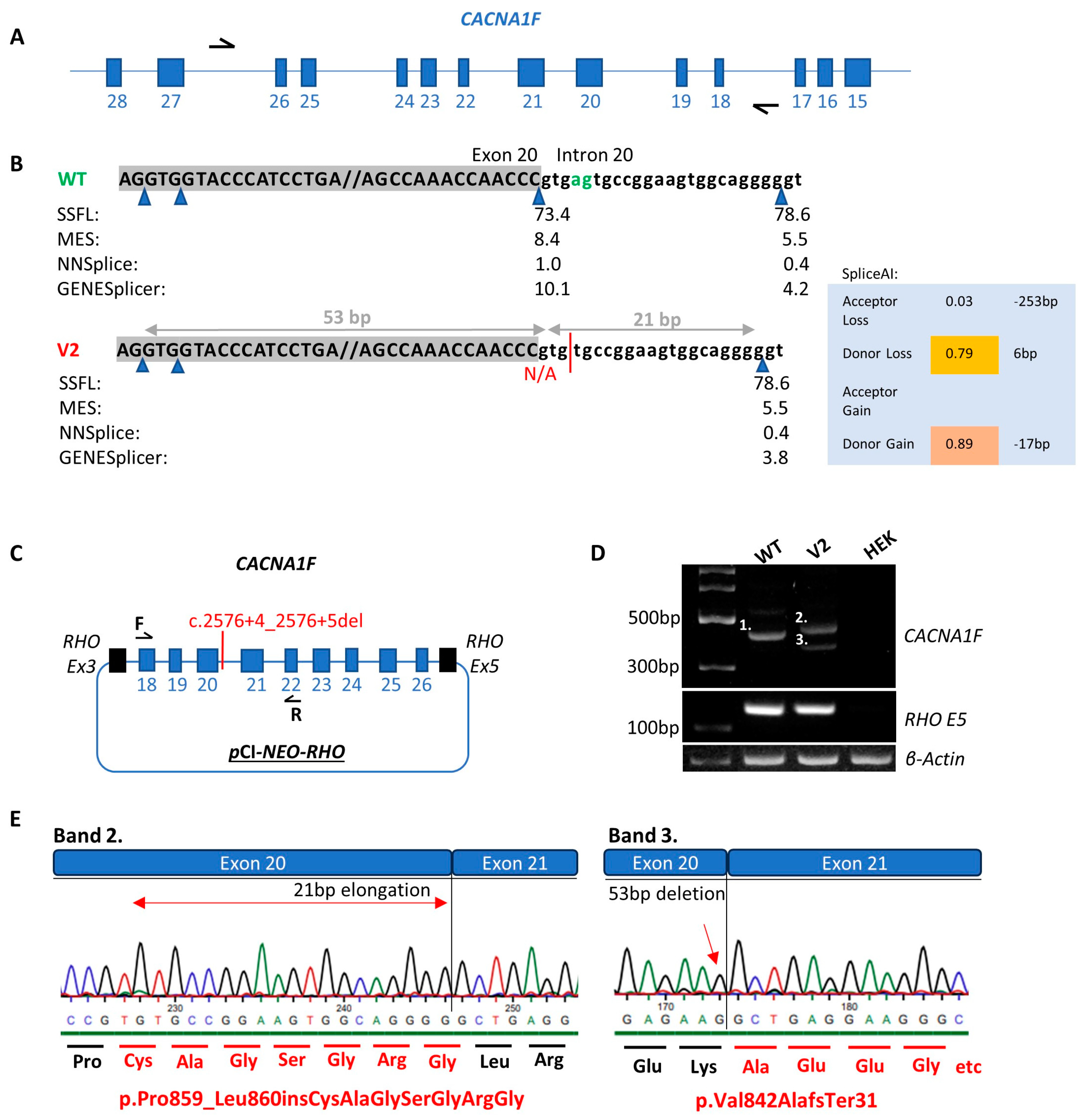
3.2. CACNA1F NM_005183.4:c.2576+4_2576+5del
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waldock, W.J.; Taylor, L.J.; Sperring, S.; Staurenghi, F.; Martinez-Fernandez de la Camara, C.; Whitfield, J.; Clouston, P.; Yusuf, I.H.; MacLaren, R.E. A hypomorphic variant of choroideremia is associated with a novel intronic mutation that leads to exon skipping. Ophthalmic Genet. 2024, 45, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Gocuk, S.A.; Lancaster, J.; Su, S.; Jolly, J.K.; Edwards, T.L.; Hickey, D.G.; Ritchie, M.E.; Blewitt, M.E.; Ayton, L.N.; Gouil, Q. Measuring X-Chromosome inactivation skew for X-linked diseases with adaptive nanopore sequencing. Genome Res. 2024, 34, 1954–1965. [Google Scholar] [CrossRef]
- Cremers, F.P.; Brunsmann, F.; van de Pol, T.J.; Pawlowitzki, I.H.; Paulsen, K.; Wieringa, B.; Ropers, H.H. Deletion of the DXS165 locus in patients with classical choroideremia. Clin. Genet. 1987, 32, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Seabra, M.C.; Brown, M.S.; Goldstein, J.L. Retinal degeneration in choroideremia: Deficiency of rab geranylgeranyl transferase. Science 1993, 259, 377–381. [Google Scholar] [CrossRef]
- Raeker, M.; Perera, N.D.; Karoukis, A.J.; Chen, L.; Feathers, K.L.; Ali, R.R.; Thompson, D.A.; Fahim, A.T. Reduced Retinal Pigment Epithelial Autophagy Due to Loss of Rab12 Prenylation in a Human iPSC-RPE Model of Choroideremia. Cells 2024, 13, 1068. [Google Scholar] [CrossRef] [PubMed]
- Daiger, S.P.; Rossiter, B.J.F.; Greenberg, J.; Christoffels, A.; Hide, W. Data Services and Software for Identifying Genes and Mutations Causing Retinal Degeneration. 1998. Available online: https://web.sph.uth.edu/RetNet/help.htm?csrt=9550839127900483002#q4 (accessed on 25 April 2024).
- Hamel, C.P. Cone rod dystrophies. Orphanet J. Rare Dis. 2007, 2, 7. [Google Scholar] [CrossRef]
- Jalkanen, R.; Mäntyjärvi, M.; Tobias, R.; Isosomppi, J.; Sankila, E.M.; Alitalo, T.; Bech-Hansen, N.T. X linked cone-rod dystrophy, CORDX3, is caused by a mutation in the CACNA1F gene. J. Med. Genet. 2006, 43, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Strom, T.M.; Nyakatura, G.; Apfelstedt-Sylla, E.; Hellebrand, H.; Lorenz, B.; Weber, B.H.; Wutz, K.; Gutwillinger, N.; Rüther, K.; Drescher, B.; et al. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998, 19, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Jalkanen, R.; Bech-Hansen, N.T.; Tobias, R.; Sankila, E.M.; Mäntyjärvi, M.; Forsius, H.; de la Chapelle, A.; Alitalo, T. A novel CACNA1F gene mutation causes Aland Island eye disease. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2498–2502. [Google Scholar] [CrossRef] [PubMed]
- Bech-Hansen, N.T.; Naylor, M.J.; Maybaum, T.A.; Pearce, W.G.; Koop, B.; Fishman, G.A.; Mets, M.; Musarella, M.A.; Boycott, K.M. Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998, 19, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Sallah, S.R.; Sergouniotis, P.I.; Barton, S.; Ramsden, S.; Taylor, R.L.; Safadi, A.; Kabir, M.; Ellingford, J.M.; Lench, N.; Lovell, S.C.; et al. Using an integrative machine learning approach utilising homology modelling to clinically interpret genetic variants: CACNA1F as an exemplar. Eur. J. Hum. Genet. 2020, 28, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, U.; Méjécase, C.; Ali, S.M.A.; Moosajee, M.; Kozak, I. A Novel Splice-Site Variant in CACNA1F Causes a Phenotype Synonymous with Åland Island Eye Disease and Incomplete Congenital Stationary Night Blindness. Genes 2021, 12, 171. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Mahroo, O.A. Negative electroretinograms: Genetic and acquired causes, diagnostic approaches and physiological insights. Eye 2021, 35, 2419–2437. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.; Cooper, D.N. The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef]
- Krawczak, M.; Thomas, N.S.; Hundrieser, B.; Mort, M.; Wittig, M.; Hampe, J.; Cooper, D.N. Single base-pair substitutions in exon-intron junctions of human genes: Nature, distribution, and consequences for mRNA splicing. Hum. Mutat. 2007, 28, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Jang, W.; Park, J.; Chae, H.; Kim, M. Comparison of In Silico Tools for Splice-Altering Variant Prediction Using Established Spliceogenic Variants: An End-User’s Point of View. Int. J. Genom. 2022, 2022, 5265686. [Google Scholar] [CrossRef]
- Wai, H.A.; Lord, J.; Lyon, M.; Gunning, A.; Kelly, H.; Cibin, P.; Seaby, E.G.; Spiers-Fitzgerald, K.; Lye, J.; Ellard, S.; et al. Blood RNA analysis can increase clinical diagnostic rate and resolve variants of uncertain significance. Genet. Med. 2020, 22, 1005–1014. [Google Scholar] [CrossRef]
- Rowlands, C.; Thomas, H.B.; Lord, J.; Wai, H.A.; Arno, G.; Beaman, G.; Sergouniotis, P.; Gomes-Silva, B.; Campbell, C.; Gossan, N.; et al. Comparison of in silico strategies to prioritize rare genomic variants impacting RNA splicing for the diagnosis of genomic disorders. Sci. Rep. 2021, 11, 20607. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Ellard, S.; Baple, E.; Berry, I.; Forrester, N.; Turnbull, C.; Owens, M.; Eccles, D.; Abbs, S.; Scott, R.; Deans, Z.; et al. ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020; Association for Clinical Genomic Science: Edinburgh, UK, 2020. [Google Scholar]
- Durkie, M.; Cassify, E.-J.; Berry, I.; Owens, M.; Turnbull, C.; Scott, R.H.; Taylor, R.W.; Deans, Z.C.; Ellard, S.; Baple, E.L.; et al. ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2024. Available online: https://www.acgs.uk.com/quality/best-practice-guidelines/ (accessed on 25 April 2024).
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. ClinGen—The Clinical Genome Resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef]
- Walker, L.C.; Hoya, M.; Wiggins, G.A.R.; Lindy, A.; Vincent, L.M.; Parsons, M.T.; Canson, D.M.; Bis-Brewer, D.; Cass, A.; Tchourbanov, A.; et al. Using the ACMG/AMP framework to capture evidence related to predicted and observed impact on splicing: Recommendations from the ClinGen SVI Splicing Subgroup. Am. J. Hum. Genet. 2023, 110, 1046–1067. [Google Scholar] [CrossRef] [PubMed]
- Fadaie, Z.; Whelan, L.; Dockery, A.; Li, C.H.Z.; van den Born, L.I.; Hoyng, C.B.; Gilissen, C.; Corominas, J.; Rowlands, C.; Megaw, R.; et al. BBS1 branchpoint variant is associated with non-syndromic retinitis pigmentosa. J. Med. Genet. 2022, 59, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Fadaie, Z.; Whelan, L.; Ben-Yosef, T.; Dockery, A.; Corradi, Z.; Gilissen, C.; Haer-Wigman, L.; Corominas, J.; Astuti, G.D.N.; de Rooij, L.; et al. Whole genome sequencing and in vitro splice assays reveal genetic causes for inherited retinal diseases. NPJ Genom. Med. 2021, 6, 97. [Google Scholar] [CrossRef] [PubMed]
- Weisschuh, N.; Buena-Atienza, E.; Wissinger, B. Splicing mutations in inherited retinal diseases. Prog. Retin. Eye Res. 2021, 80, 100874. [Google Scholar] [CrossRef]
- Stephenson, K.A.J.; Zhu, J.; Wynne, N.; Dockery, A.; Cairns, R.M.; Duignan, E.; Whelan, L.; Malone, C.P.; Dempsey, H.; Collins, K.; et al. Target 5000: A standardized all-Ireland pathway for the diagnosis and management of inherited retinal degenerations. Orphanet J. Rare Dis. 2021, 16, 200. [Google Scholar] [CrossRef]
- Dockery, A.; Stephenson, K.; Keegan, D.; Wynne, N.; Silvestri, G.; Humphries, P.; Kenna, P.F.; Carrigan, M.; Farrar, G.J. Target 5000: Target Capture Sequencing for Inherited Retinal Degenerations. Genes 2017, 8, 304. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Fokkema, I.; Kroon, M.; López Hernández, J.A.; Asscheman, D.; Lugtenburg, I.; Hoogenboom, J.; den Dunnen, J.T. The LOVD3 platform: Efficient genome-wide sharing of genetic variants. Eur. J. Hum. Genet. 2021, 29, 1796–1803. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Firth, H.V.; Richards, S.M.; Bevan, A.P.; Clayton, S.; Corpas, M.; Rajan, D.; Van Vooren, S.; Moreau, Y.; Pettett, R.M.; Carter, N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009, 84, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Uniprot Consortium. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.B.; Senapathy, P. RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987, 15, 7155–7174. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Lin, X.; Salzberg, S.L. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 2001, 29, 1185–1190. [Google Scholar] [CrossRef]
- de Sainte Agathe, J.M.; Filser, M.; Isidor, B.; Besnard, T.; Gueguen, P.; Perrin, A.; Van Goethem, C.; Verebi, C.; Masingue, M.; Rendu, J.; et al. SpliceAI-visual: A free online tool to improve SpliceAI splicing variant interpretation. Hum. Genom. 2023, 17, 7. [Google Scholar] [CrossRef] [PubMed]
- Sangermano, R.; Bax, N.M.; Bauwens, M.; van den Born, L.I.; De Baere, E.; Garanto, A.; Collin, R.W.; Goercharn-Ramlal, A.S.; den Engelsman-van Dijk, A.H.; Rohrschneider, K.; et al. Photoreceptor Progenitor mRNA Analysis Reveals Exon Skipping Resulting from the ABCA4 c.5461-10T→C Mutation in Stargardt Disease. Ophthalmology 2016, 123, 1375–1385. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Coban-Akdemir, Z.; White, J.J.; Song, X.; Jhangiani, S.N.; Fatih, J.M.; Gambin, T.; Bayram, Y.; Chinn, I.K.; Karaca, E.; Punetha, J.; et al. Identifying Genes Whose Mutant Transcripts Cause Dominant Disease Traits by Potential Gain-of-Function Alleles. Am. J. Hum. Genet. 2018, 103, 171–187. [Google Scholar] [CrossRef]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. InterPro in 2022. Nucleic Acids Res. 2023, 51, D418–D427. [Google Scholar] [CrossRef]
- Pylypenko, O.; Rak, A.; Reents, R.; Niculae, A.; Sidorovitch, V.; Cioaca, M.D.; Bessolitsyna, E.; Thomä, N.H.; Waldmann, H.; Schlichting, I.; et al. Structure of Rab escort protein-1 in complex with Rab geranylgeranyltransferase. Mol. Cell 2003, 11, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Rak, A.; Pylypenko, O.; Niculae, A.; Pyatkov, K.; Goody, R.S.; Alexandrov, K. Structure of the Rab7:REP-1 complex: Insights into the mechanism of Rab prenylation and choroideremia disease. Cell 2004, 117, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Wutz, K.; Sauer, C.; Zrenner, E.; Lorenz, B.; Alitalo, T.; Broghammer, M.; Hergersberg, M.; de la Chapelle, A.; Weber, B.H.; Wissinger, B.; et al. Thirty distinct CACNA1F mutations in 33 families with incomplete type of XLCSNB and Cacna1f expression profiling in mouse retina. Eur. J. Hum. Genet. 2002, 10, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Albert, S.; Garanto, A.; Sangermano, R.; Khan, M.; Bax, N.M.; Hoyng, C.B.; Zernant, J.; Lee, W.; Allikmets, R.; Collin, R.W.J.; et al. Identification and Rescue of Splice Defects Caused by Two Neighboring Deep-Intronic ABCA4 Mutations Underlying Stargardt Disease. Am. J. Hum. Genet. 2018, 102, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Wang, P. Phenotype and genotype of CACNA1F related diseases. Investig. Ophthalmol. Vis. Sci. 2024, 65, 5267. [Google Scholar]
- Vincent, A.; Wright, T.; Day, M.A.; Westall, C.A.; Héon, E. A novel p.Gly603Arg mutation in CACNA1F causes Åland island eye disease and incomplete congenital stationary night blindness phenotypes in a family. Mol. Vis. 2011, 17, 3262–3270. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Family ID | Status | Sex | Age of Onset | First Symptoms | BCVA | Object V Horizontal Visual Field Remaining (Goldman) | Lens | Colour Vision | Rod Response (RE) | Rod Response (LE) | Oscillatory Potential (RE) | Oscillatory Potential (LE) | Photopic (RE) | Photopic (LE) | 30 Hz Flicker (RE) | 30 Hz Flicker (LE) | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pt-1 | A | Affected | Male | 11 | Nyctalopia, Photophobia, RE worse than LE | RE PL, LE 6/60 age 47 | RE NR, LE 20° age 47 | Subcapsular Bilateral Cataracts Age 38 | Pathologic Colour Discrimination | NR | NR | Reduced and Delayed Age 20 | Reduced and Delayed Age 20 | Significantly Reduced and Delayed Age 20 | Significantly Reduced and Delayed Age 20 | Significantly Reduced and Delayed Age 20 | Significantly Reduced and Delayed Age 20 | RE Extropia |
| Pt-4 | A | Carrier | Female | N/A | None, Obligate Carrier | RE 6/4.8, LE 6/7.5 | RE 140°, LE 135° age 19 | No Cataracts | RE Diffuse Colour Error, LE Normal | Within Normal limits | Within Normal limits | N/A | N/A | Within Normal limits | Within Normal limits | N/A | N/A | RPE Changes Consistent with Obligate X-Linked CHM carrier |
| Pt-5 | A | Affected | Male | 6 | Nyctalopia, Photophobia | RE 6/7.5, LE 6/7.5 | RE 130°, LE 130° age 16 | Cells in Vitreous Body | RE Normal, LE Diffuse Colour Error | Significantly Reduced Age 16 | Significantly Reduced Age 16 | NR age 16 | NR age 16 | Delayed and Significantly Reduced Age 16 | Delayed and Significantly Reduced Age 16 | Significantly Reduced Age 16 | Significantly Reduced Age 16 | N/A |
| Pt-6 | B | Affected | Male | 20 | Nyctalopia | RE 3/60, LE 6/24 Age 50 Years | BE < 5° | No cataracts | Pathologic Colour Discrimination onset Age 40 | NR | NR | N/A | N/A | Reduced | Reduced | Delayed and Reduced | Delayed and Reduced | Abnormal EOG |
| Pt-7 | C | Affected | Male | 25 | Glare | RE 6/60, LE CF Age 64 Years | BE 20° | Bilateral Cataracts Age of Onset 65 Years | Normal colour Discrimination | Borderline Reduced | Borderline Reduced | N/A | N/A | Reduced | Reduced | NR | NR | Myopic, congenital nystagmus, VEP significantly delayed and borderline reduced |
| Variant | Population Data | Computational Data | Segregation Data | Other Data | Points | Classification |
|---|---|---|---|---|---|---|
| Classification prior to midigene functional analysis | ||||||
| CHM c.941-11T>G | PM2_Supporting | PP3 | N/A | PP4 | 3 | VUS |
| CACNA1F c.2576+4_2576+5del | PM2_Supporting | PP3 | N/A | N/A | 2 | VUS |
| Classification after midigene functional analysis | ||||||
| CHM c.941-11T>G | PM2_Supporting | PVS1_Strong | N/A | PP4 | 6 | Likely Pathogenic |
| CACNA1F c.2576+4_2576+5del | PM2_Supporting | PVS1_Strong | N/A | N/A | 5 | Hot VUS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ridgeway, A.R.; Shortall, C.; Finnegan, L.K.; Long, R.; Matthews, E.; Dockery, A.; Kopčić, E.; Whelan, L.; Kirk, C.; Silvestri, G.; et al. Novel Splice-Altering Variants in the CHM and CACNA1F Genes Causative of X-Linked Choroideremia and Cone Dystrophy. Genes 2025, 16, 25. https://doi.org/10.3390/genes16010025
Ridgeway AR, Shortall C, Finnegan LK, Long R, Matthews E, Dockery A, Kopčić E, Whelan L, Kirk C, Silvestri G, et al. Novel Splice-Altering Variants in the CHM and CACNA1F Genes Causative of X-Linked Choroideremia and Cone Dystrophy. Genes. 2025; 16(1):25. https://doi.org/10.3390/genes16010025
Chicago/Turabian StyleRidgeway, Anna R., Ciara Shortall, Laura K. Finnegan, Róisín Long, Evan Matthews, Adrian Dockery, Ella Kopčić, Laura Whelan, Claire Kirk, Giuliana Silvestri, and et al. 2025. "Novel Splice-Altering Variants in the CHM and CACNA1F Genes Causative of X-Linked Choroideremia and Cone Dystrophy" Genes 16, no. 1: 25. https://doi.org/10.3390/genes16010025
APA StyleRidgeway, A. R., Shortall, C., Finnegan, L. K., Long, R., Matthews, E., Dockery, A., Kopčić, E., Whelan, L., Kirk, C., Silvestri, G., Turner, J., Keegan, D. J., Millington-Ward, S., Chadderton, N., Duignan, E., Kenna, P. F., & Farrar, G. J. (2025). Novel Splice-Altering Variants in the CHM and CACNA1F Genes Causative of X-Linked Choroideremia and Cone Dystrophy. Genes, 16(1), 25. https://doi.org/10.3390/genes16010025