
A Case of Non-Syndromic Congenital Cataracts Caused by a Novel MAF Variant in the C-Terminal DNA-Binding Domain—Case Report and Literature Review

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Information and History
2.2. Genetic Testing
3. Results
3.1. Exome Sequencing
3.2. Literature Review
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shiels, A.; Hejtmancik, J.F. Mutations and mechanisms in congenital and age-related cataracts. Exp. Eye Res. 2017, 156, 95–102. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Shiels, A.; Bennett, T.M.; Hejtmancik, J.F. Cat-Map: Putting cataract on the map. Mol. Vis. 2010, 16, 2007–2015. [Google Scholar] [PubMed]
- Rossen, J.L.; Bohnsack, B.L.; Zhang, K.X.; Ing, A.; Drackley, A.; Castelluccio, V.; Ralay-Ranaivo, H. Evaluation of Genetic Testing in a Cohort of Diverse Pediatric Patients in the United States with Congenital Cataracts. Genes 2023, 14, 608. [Google Scholar] [CrossRef] [PubMed]
- Franklin by Genoox: The Future of Genomic Medicine. 2023. Available online: https://franklin.genoox.com (accessed on 1 July 2023).
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Narumi, Y.; Nishina, S.; Tokimitsu, M.; Aoki, Y.; Kosaki, R.; Wakui, K.; Azuma, N.; Murata, T.; Takada, F.; Fukushima, Y.; et al. Identification of a novel missense mutation of MAF in a Japanese family with congenital cataract by whole exome sequencing: A clinical report and review of literature. Am. J. Med. Genet. A 2014, 164A, 1272–1276. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, R.V.; Munier, F.; Balmer, A.; Farrar, N.; Perveen, R.; Black, G.C. Pulverulent cataract with variably associated microcornea and iris coloboma in a MAF mutation family. Br. J. Ophthalmol. 2003, 87, 411–412. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.; Malka, S.; Harding, P.; Palma, J.; Dunbar, H.; Moosajee, M. Molecular diagnostic challenges for non-retinal developmental eye disorders in the United Kingdom. Am. J. Med. Genet. C Semin Med. Genet. 2020, 184, 578–589. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dudakova, L.; Stranecky, V.; Ulmanova, O.; Hlavova, E.; Trková, M.; Vincent, A.L.; Liskova, P. Segregation of a novel p.(Ser270Tyr) MAF mutation and p.(Tyr56∗) CRYGD variant in a family with dominantly inherited congenital cataracts. Mol. Biol. Rep. 2017, 44, 435–440, Erratum in Mol. Biol. Rep. 2017, 44, 441. [Google Scholar] [CrossRef] [PubMed]
- Si, N.; Song, Z.; Meng, X.; Li, X.; Xiao, W.; Zhang, X. A novel MAF missense mutation leads to congenital nuclear cataract by impacting the transactivation of crystallin and noncrystallin genes. Gene 2019, 692, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.S.; Grigg, J.R.; Ho, G.; Prokudin, I.; Farnsworth, E.; Holman, K.; Cheng, A.; Billson, F.A.; Martin, F.; Fraser, C.; et al. Sporadic and Familial Congenital Cataracts: Mutational Spectrum and New Diagnoses Using Next-Generation Sequencing. Hum. Mutat. 2016, 37, 371–384. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jamieson, R.V.; Perveen, R.; Kerr, B.; Carette, M.; Yardley, J.; Heon, E.; Wirth, M.G.; van Heyningen, V.; Donnai, D.; Munier, F.; et al. Domain disruption and mutation of the bZIP transcription factor, MAF, associated with cataract, ocular anterior segment dysgenesis and coloboma. Hum. Mol. Genet. 2002, 11, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Xiao, X.; Li, S.; Guo, X.; Zhang, Q. Exome Sequencing of 18 Chinese Families with Congenital Cataracts: A New Sight of the NHS Gene. PLoS ONE 2014, 9, e100455. [Google Scholar] [CrossRef]
- Vanita, V.; Singh, D.; Robinson, P.N.; Sperling, K.; Singh, J.R. A novel mutation in the DNA-binding domain of MAF at 16q23.1 associated with autosomal dominant “cerulean cataract” in an Indian family. Am. J. Med. Genet. A 2006, 140, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Hayward, J.D.; Tailor, V.; Nyanhete, R.; Ahlfors, H.; Gabriel, C.; Jannini, T.B.; Abbou-Rayyah, Y.; Henderson, R.; Nischal, K.K.; et al. The Oculome Panel Test: Next-Generation Sequencing to Diagnose a Diverse Range of Genetic Developmental Eye Disorders. Ophthalmology 2019, 126, 888–907. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.; Eiberg, H.; Rosenberg, T. Novel MAF mutation in a family with congenital cataract-microcornea syndrome. Mol. Vis. 2007, 13, 2019–2022. [Google Scholar] [PubMed]
- Rechsteiner, D.; Issler, L.; Koller, S.; Lang, E.; Bähr, L.; Feil, S.; Rüegger, C.M.; Kottke, R.; Toelle, S.P.; Zweifel, N.; et al. Genetic Analysis in a Swiss Cohort of Bilateral Congenital Cataract. JAMA Ophthalmol. 2021, 139, 691–700. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ma, A.; Grigg, J.R.; Flaherty, M.; Smith, J.; Minoche, A.E.; Cowley, M.J.; Nash, B.M.; Ho, G.; Gayagay, T.; Lai, T.; et al. Genome sequencing in congenital cataracts improves diagnostic yield. Hum. Mutat. 2021, 42, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Qin, T.; Tan, H.; Ding, X.; Lin, X.; Li, J.; Lin, Z.; Sun, L.; Lin, H.; Chen, W. Broadening the genotypic and phenotypic spectrum of MAF in three Chinese Han congenital cataracts families. Am. J. Med. Genet. A 2022, 188, 2888–2898. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Leng, Y.; Han, S.; Yan, L.; Lu, C.; Luo, Y.; Zhang, X.; Cao, L. Clinical and genetic characteristics of Chinese patients with familial or sporadic pediatric cataract. Orphanet. J. Rare Dis. 2018, 13, 94. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.; Mikkelsen, A.; Nürnberg, P.; Nürnberg, G.; Anjum, I.; Eiberg, H.; Rosenberg, T. Comprehensive Mutational Screening in a Cohort of Danish Families with Hereditary Congenital Cataract. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3291–3303. [Google Scholar] [CrossRef]
- Kawauchi, S.; Takahashi, S.; Nakajima, O.; Ogino, H.; Morita, M.; Nishizawa, M.; Yasuda, K.; Yamamoto, M. Regulation of lens fiber cell differentiation by transcription factor c-Maf. J. Biol. Chem. 1999, 274, 19254–19260. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Serria, M.S.; Ikeda, H.; Yoshida, K.; Imaki, J.; Nishi, S. Regulation of c-Maf gene expression by Pax6 in cultured cells. Nucleic Acids Res. 2001, 29, 1228–1237. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, Q.; Dowhan, D.H.; Liang, D.; Moore, D.D.; Overbeek, P.A. CREB-binding protein/p300 co-activation of crystallin gene expression. J. Biol. Chem. 2002, 277, 24081–24089. [Google Scholar] [CrossRef] [PubMed]
- Lyon, M.F.; Jamieson, R.V.; Perveen, R.; Glenister, P.H.; Griffiths, R.; Boyd, Y.; Glimcher, L.H.; Favor, J.; Munier, F.L.; Black, G.C.M. A dominant mutation within the DNA-binding domain of the bZIP transcription factor Maf causes murine cataract and results in selective alteration in DNA binding. Hum. Mol. Genet. 2003, 12, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Perveen, R.; Favor, J.; Jamieson, R.V.; Ray, D.W.; Black, G.C. A heterozygous c-Maf transactivation domain mutation causes congenital cataract and enhances target gene activation. Hum. Mol. Genet. 2007, 16, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, S.; Ronda, L.; Annoni, C.; Contini, A.; Erba, E.; Gelmi, M.L.; Piano, R.; Paredi, G.; Mozzarelli, A.; Bettati, S. Molecular insights into dimerization inhibition of c-Maf transcription factor. Biochim. Biophys. Acta 2014, 1844, 2108–2115. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Guanga, G.; Wan, C.; Rose, R. A Novel DNA Binding Mechanism for Maf Basic Region-Leucine Zipper Factors Inferred from a MafA–DNA Complex Structure and Binding Specificities. Biochemistry 2012, 51, 9706–9717. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Yun, J.; Li, Z.K.; Xu, C.T.; Pan, B.R. Epidemiology and molecular genetics of congenital cataracts. Int. J. Ophthalmol. 2011, 4, 422–432. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Javadiyan, S.; Craig, J.E.; Sharma, S.; Lower, K.M.; Casey, T.; Haan, E.; Souzeau, E.; Burdon, K.P. Novel missense mutation in the bZIP transcription factor, MAF, associated with congenital cataract, developmental delay, seizures and hearing loss (Aymé-Gripp syndrome). BMC Med. Genet. 2017, 18, 52. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Aymé, S.; Philip, N. Fine-Lubinsky syndrome: A fourth patient with brachycephaly, deafness, cataract, microstomia and mental retardation. Clin. Dysmorphol. 1996, 5, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Gripp, K.W.; Nicholson, L.; Scott, C.I., Jr. Apparently new syndrome of congenital cataracts, sensorineural deafness, Down syndrome-like facial appearance, short stature, and mental retardation. Am. J. Med. Genet. 1996, 61, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Ring, B.Z.; Cordes, S.P.; Overbeek, P.A.; Barsh, G.S. Regulation of mouse lens fiber cell development and differentiation by the Maf gene. Development 2000, 127, 307–317. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef] [PubMed]
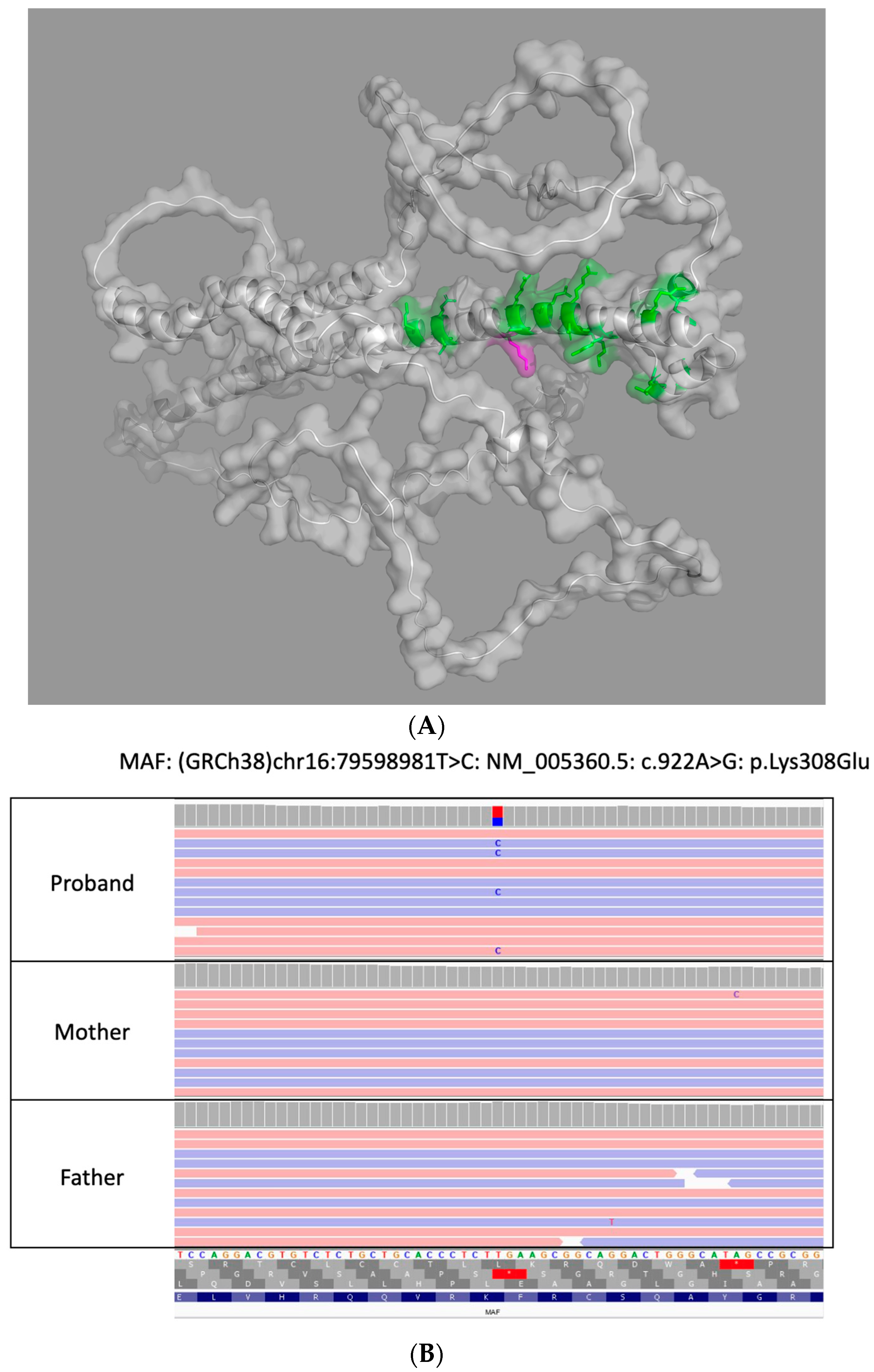

{kind=link}
{kind=link}
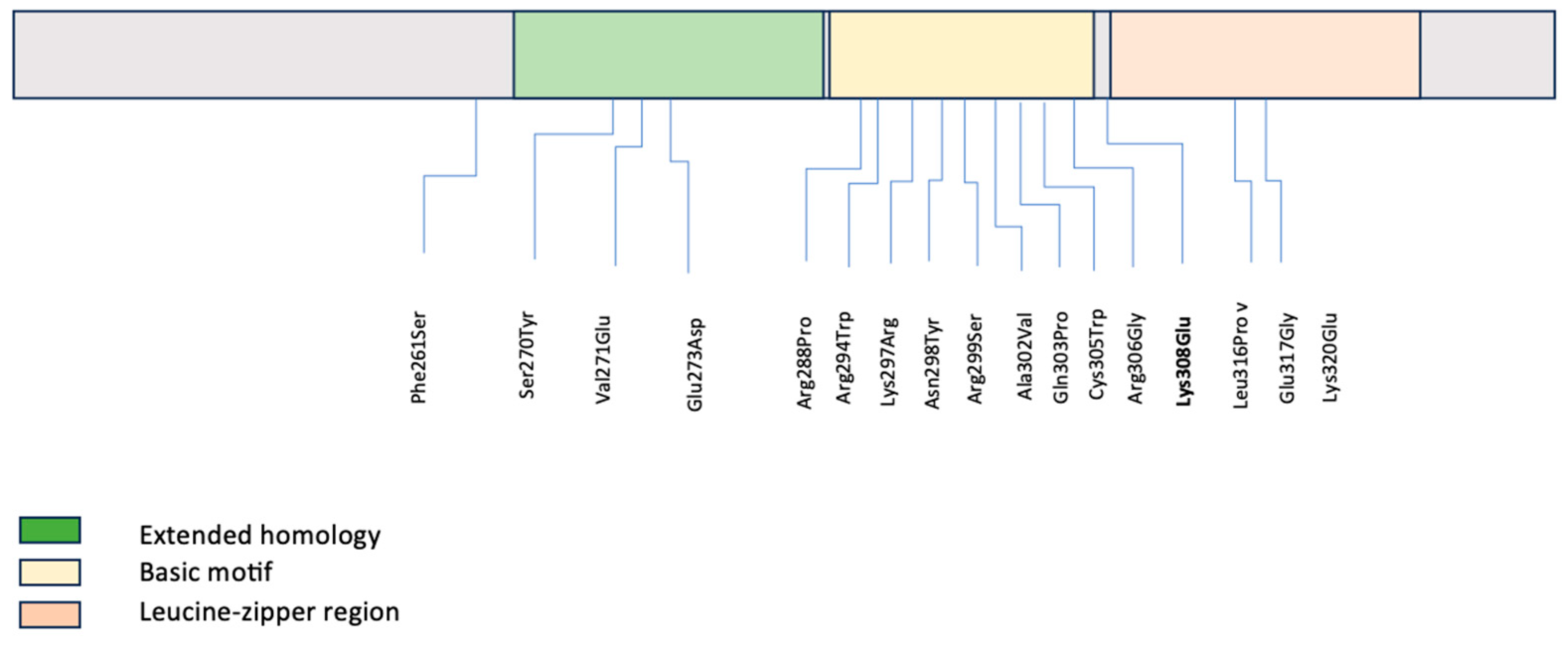
| Original Report of MAF Variant | cDNA Variant | Protein Change | Protein Domain | Variant Type | Cataract Phenotype | Anterior Segment Manifestations | Secondary Glaucoma | Inheritance | ACMG Classification | Citations |
|---|---|---|---|---|---|---|---|---|---|---|
| Jackson 2020 | c.782T>C | Phe261Ser | N/A | Missense | N/A | N/A | No | AD | N/A | [10] |
| Dudakova 2017 | c.809C>A | Ser270Tyr | Extended homology region | Missense | Nuclear cataract | Bilateral microcornea | Yes | AD | P | [11] |
| Si 2019 | c.812T>A | Val271Glu | Extended homology region | Missense | Nuclear cataracts with lamellar opacities | N/A | No | AD | P | [12] |
| Ma 2016 | c.819G>C | Glu273Asp | Extended homology region | Missense | N/A | N/A | No | Presumed de novo or sporadic | LP | [13] |
| Jamieson 2002 | c.863G>C | Arg288Pro | Basic motif | Missense | Family 1—cortical pulverulent cataract with anterior + posterior sutural densities | Family 1—opaque corneas, Peters anomaly | No | AD; | N/A | [14] |
| Family 2—cortical, pulverulent, lamellar lens opacities; nuclear cataract | Family 2—two individuals with microcornea, one with bilateral iris coloboma | Family 1 with unbalanced and balanced forms of translocation | ||||||||
| Sun 2014 | c.880C>T | Arg294Trp | Basic motif | Missense | Nuclear cataracts | N/A | No | AD | P | [15] |
| Ma 2016 | c.880C>T | Arg294Trp | Basic motif | Missense | N/A | N/A | No | Presumed de novo or sporadic | LP | [13] |
| Vanita 2006 | c.890A>G | Lys297Arg | Basic motif | Missense | Cerulean cataract | Microcornea | No | AD | P | [16] |
| Patel 2019 | c.892A>T | Asn298Tyr | Basic motif | Missense | N/A | N/A | No | Unknown | N/A | [17] |
| Hansen 2007 | c.895C>A | Arg299Ser | Basic motif | Missense | Lamellar (zonular) and star-shaped | Microcornea, occasional iris coloboma | No | AD | N/A | [18] |
| Rechsteiner 2021 | c.905C>T | Ala302Val | Basic motif | Missense | N/A | N/A | No | N/A | LP | [19] |
| Narumi 2014 | c.908A>C | Gln303Pro | Basic motif | Missense | Lamellar cataract | Microcornea and/or iris coloboma in some affected individuals | No | AD | N/A | [7] |
| Ma 2016 | c.915C>T | Cys305Trp | Basic motif | Missense | N/A | N/A | Yes | Presumed de novo or sporadic | LP | [13] |
| Ma 2021 | c.916C>G | Arg306Gly | Basic motif | Missense | N/A | N/A | No | Presumed de novo or sporadic | N/A | [20] |
| Wang 2022 | c.947T>C | Leu316Pro | Leucine zipper region | Missense | Nuclear, zonular, and structural cataract | N/A | No | AD | LP | [21] |
| Li 2018 | c.950A>G | Glu317Gly | Leucine zipper region | Missense | Posterior polar cataract | N/A | No | AD | P | [22] |
| Hansen 2009 | c.958A>G | Lys320Glu | Leucine zipper region | Missense | N/A | Microcornea | No | AD | N/A | [23] |
| Citation | Model(s) Used | Experimental Finding(s) | Reported Phenotype |
|---|---|---|---|
| [24] | Embryonic and adult mice heterozygous or homozygous for mutated murine c-maf gene containing a b-galactosidase (lacZ) gene insertion. | c-Maf expression is higher in primary fiber cells than epithelial cells in mice. γ-crystallin expression was not detected in c-Maf-deficient newborns, and αA-, αB-, and β-crystallins were downregulated. | Embryos homozygous for c-maf mutation have abnormal lenses with defective differentiation of lens fiber cells (no elongation). |
| [25] | c-maf knockout mouse model with abnormal lens development | Pax6 and c-Maf mRNAs are expressed in the lens equator. | N/A |
| [14] | Mouse | A translocation within Maf, t(5;16)(p15.3;q23.2), that was isolated from a family with cataract, anterior segment dysgenesis, and microphthalmia, and cloned demonstrated defective lens formation and microphthalmia in mouse embryos. | The null mutant Maf mouse embryo demonstrated defective lens formation and microphthalmia. |
| [26] | COS-1 and human lens epithelial cells (HLECB3) transfected with a reporter (one of three mouse crystallin promoter-luciferase reporters (αA, βB2, and γF) or a MARE-TK-luciferase reporter) and a plasmid encoding c-MAF, Prox-1, and/or Sox-1 in the presence of absence of CBP or p300. | c-Maf expression transactivated each of the promoters. Coexpression of CBP or p300 with c-MAF synergistically co-activated each promoter. c-Maf likely upregulates crystallin gene expression by recruiting CBP and/or p300 to crystallin promoters. | N/A |
| [27] | Ofl mice with Maf mutation (R291Q) demonstrating cataract | The mutation in Ofl mice result in selectively altered DNA binding affinities to target oligonucleotides with downstream cascade effects. | Homozygous mice fail to differentiate lens fiber cells (remain columnar epithelium). Heterozygous Ofl mice crossed with different genetic backgrounds generate anterior segment abnormalities. |
| [28] | Murine c-Maf mutant (ENU424) associated with isolated congenital cataract. | The large Maf transactivation mutation enhances interaction with transcriptional co-activator p300. | ENU424 heterozygotes expressed a mild granular nuclear opacity, and homozygotes expressed a denser and more severe nuclear opacity. |
| [12] | HEK 293 T cells transfected with pcDNA3.1-MAF expression plasmid and pGL3-crystallin/non-crystallin promoter luciferase plasmid. Control: pRL-TK Renilla luciferase vector | Val271Glu variant (c.812T>A) significantly impaired the transactivation of four crystallin genes (CRYGA, CRYAA, CRYBA1, and CRYBA4) involved in lens composition. | Non-syndromic congenital nuclear and lamellar opacities observed in family. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, S.H.; Yap, K.L.; Allegretti, V.; Drackley, A.; Ing, A.; Gordon, A.; Skol, A.; McMullen, P.; Bohnsack, B.L.; Kurup, S.P.; et al. A Case of Non-Syndromic Congenital Cataracts Caused by a Novel MAF Variant in the C-Terminal DNA-Binding Domain—Case Report and Literature Review. Genes 2024, 15, 686. https://doi.org/10.3390/genes15060686
Zhao SH, Yap KL, Allegretti V, Drackley A, Ing A, Gordon A, Skol A, McMullen P, Bohnsack BL, Kurup SP, et al. A Case of Non-Syndromic Congenital Cataracts Caused by a Novel MAF Variant in the C-Terminal DNA-Binding Domain—Case Report and Literature Review. Genes. 2024; 15(6):686. https://doi.org/10.3390/genes15060686
Chicago/Turabian StyleZhao, Sharon H., Kai Lee Yap, Valerie Allegretti, Andy Drackley, Alexander Ing, Adam Gordon, Andrew Skol, Patrick McMullen, Brenda L. Bohnsack, Sudhi P. Kurup, and et al. 2024. "A Case of Non-Syndromic Congenital Cataracts Caused by a Novel MAF Variant in the C-Terminal DNA-Binding Domain—Case Report and Literature Review" Genes 15, no. 6: 686. https://doi.org/10.3390/genes15060686
APA StyleZhao, S. H., Yap, K. L., Allegretti, V., Drackley, A., Ing, A., Gordon, A., Skol, A., McMullen, P., Bohnsack, B. L., Kurup, S. P., Ralay Ranaivo, H., & Rossen, J. L. (2024). A Case of Non-Syndromic Congenital Cataracts Caused by a Novel MAF Variant in the C-Terminal DNA-Binding Domain—Case Report and Literature Review. Genes, 15(6), 686. https://doi.org/10.3390/genes15060686