
Neuronal Ceroid Lipofuscinosis in a Mixed-Breed Dog with a Splice Site Variant in CLN6

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
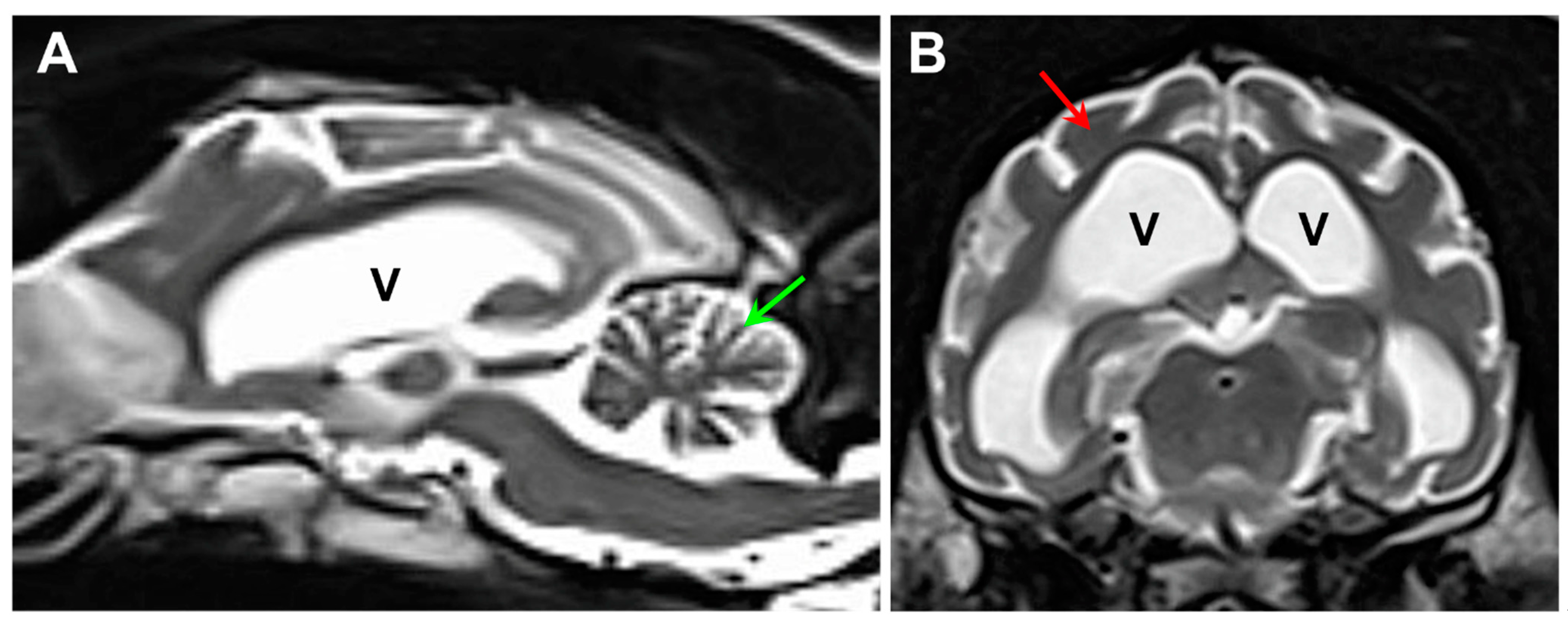
3. Results
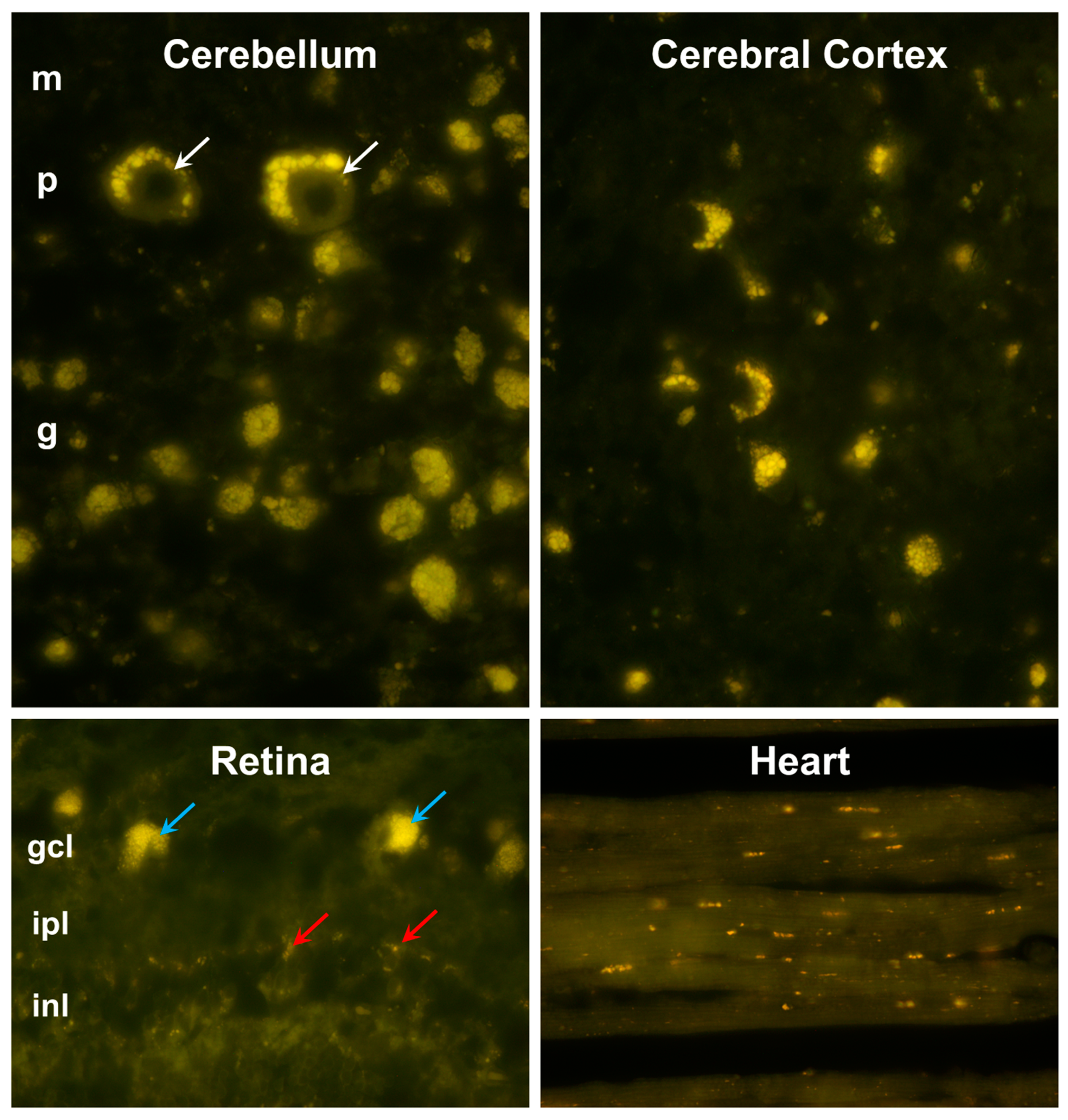
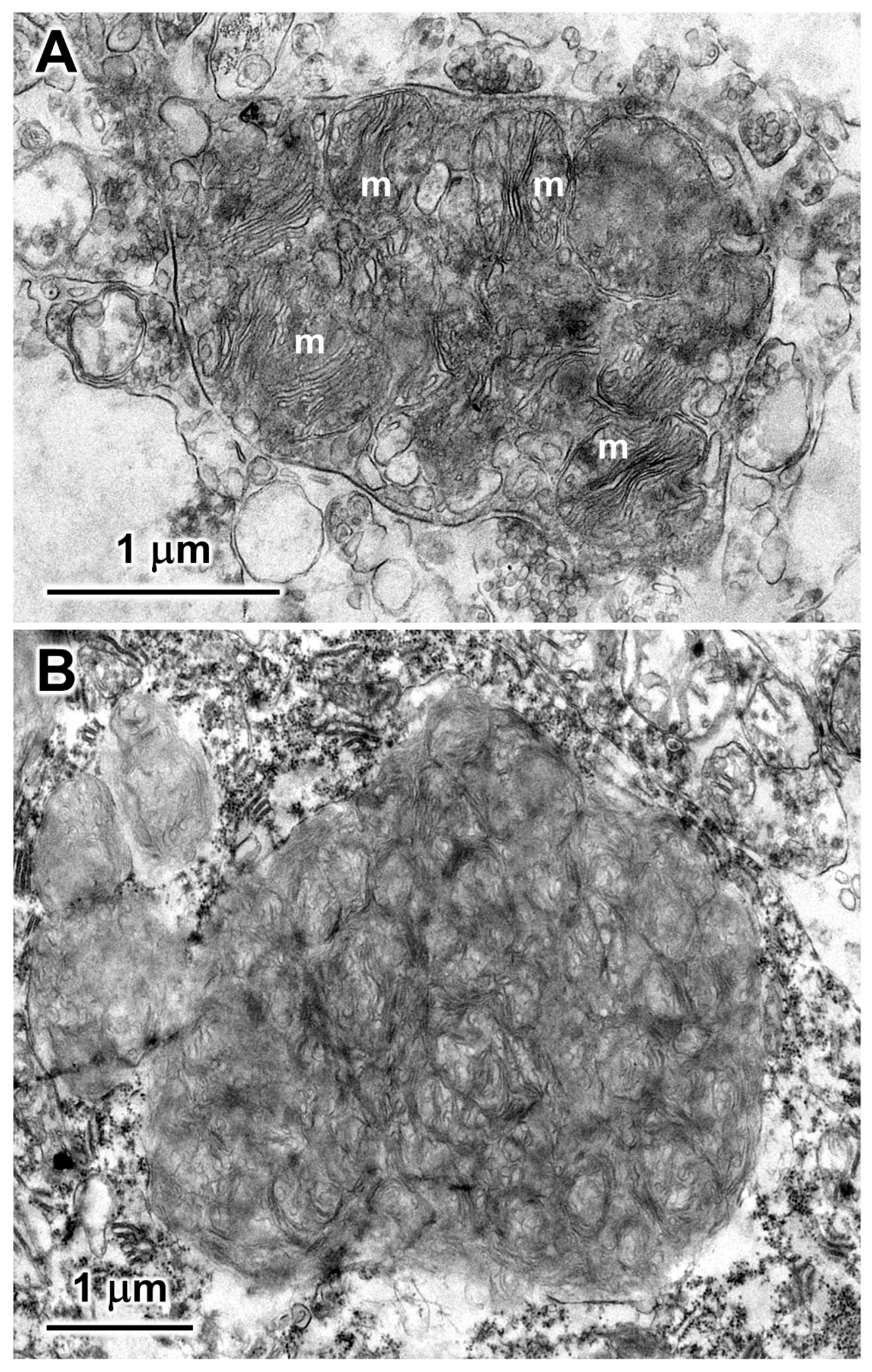
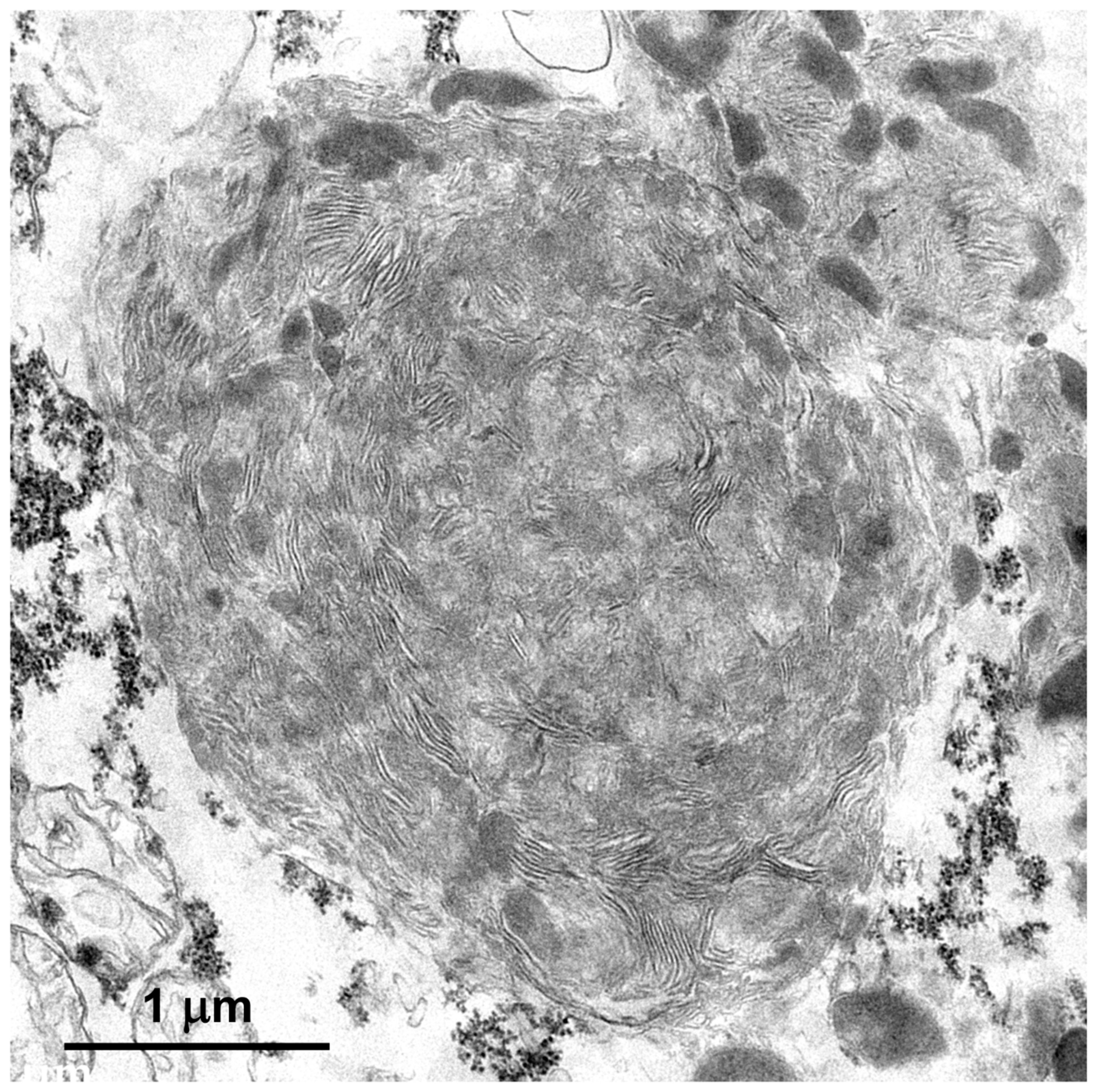
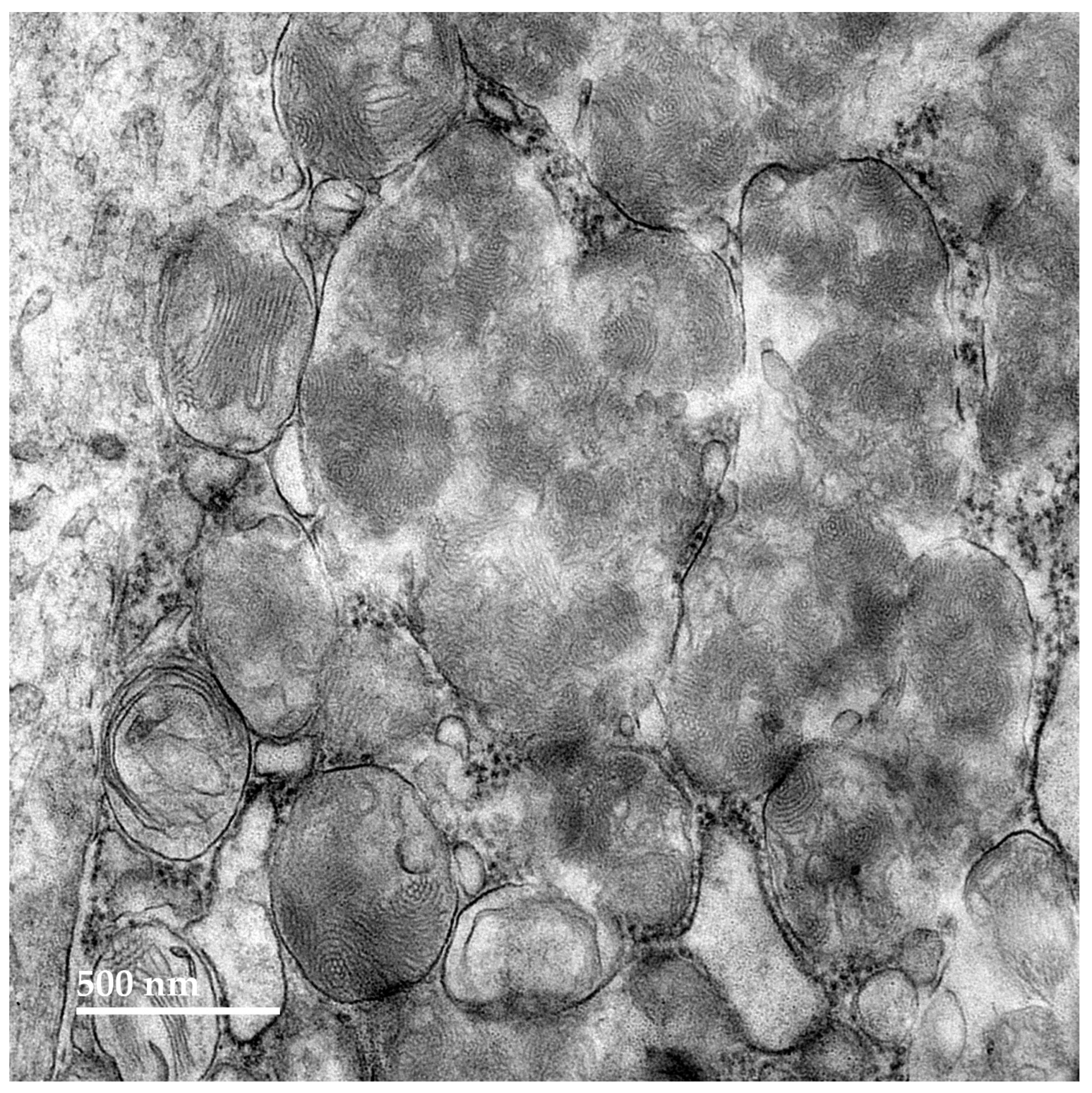
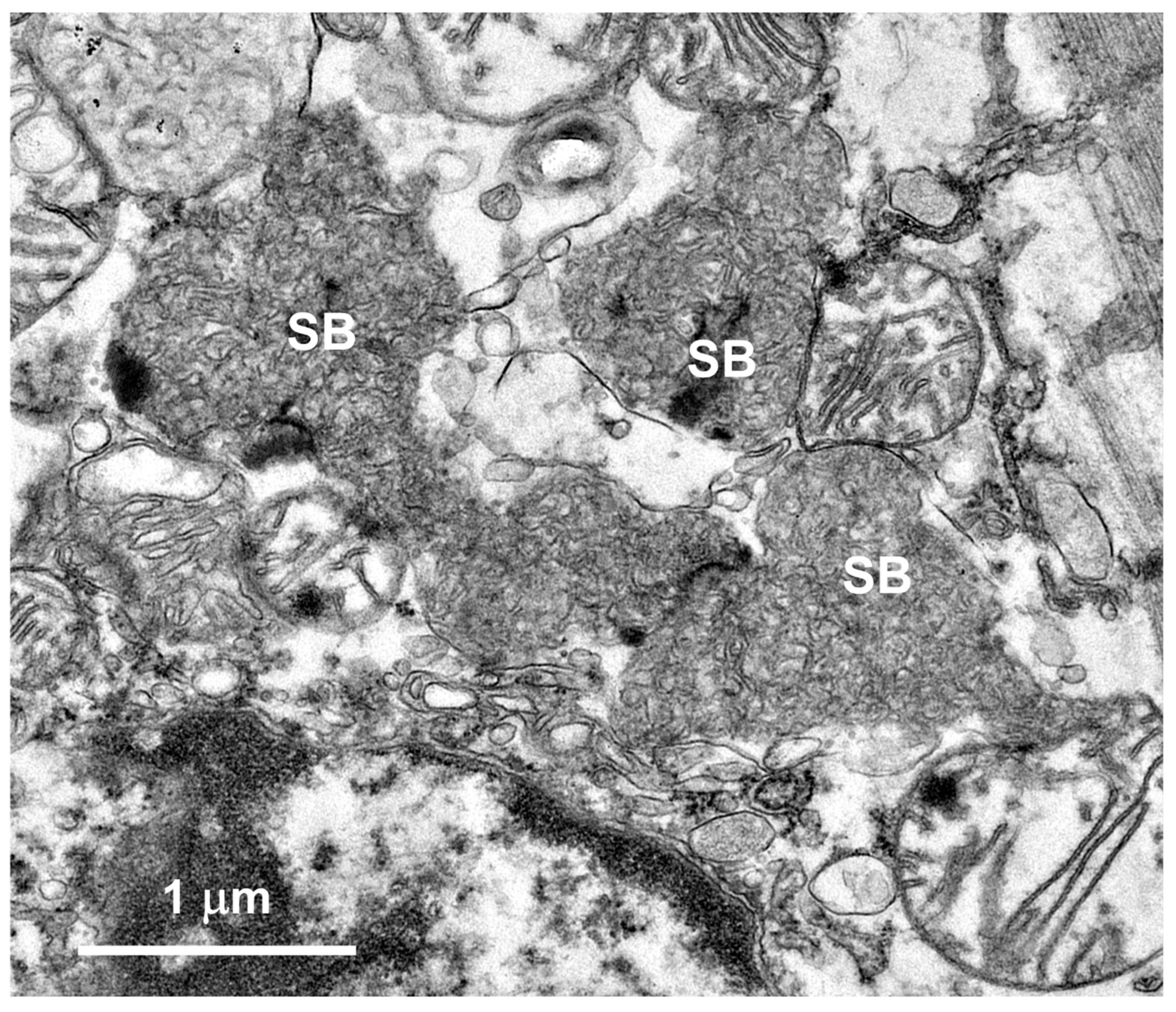
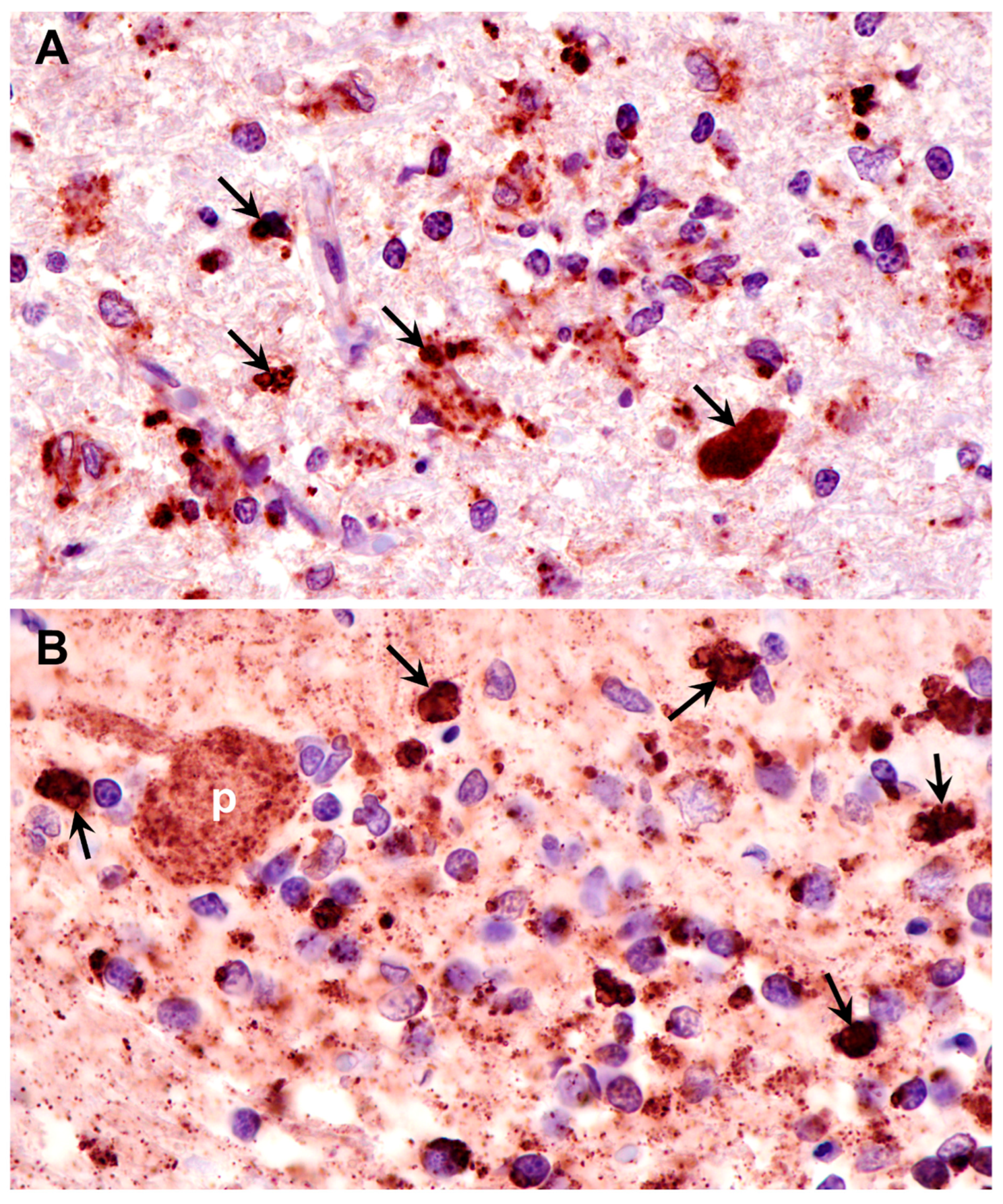
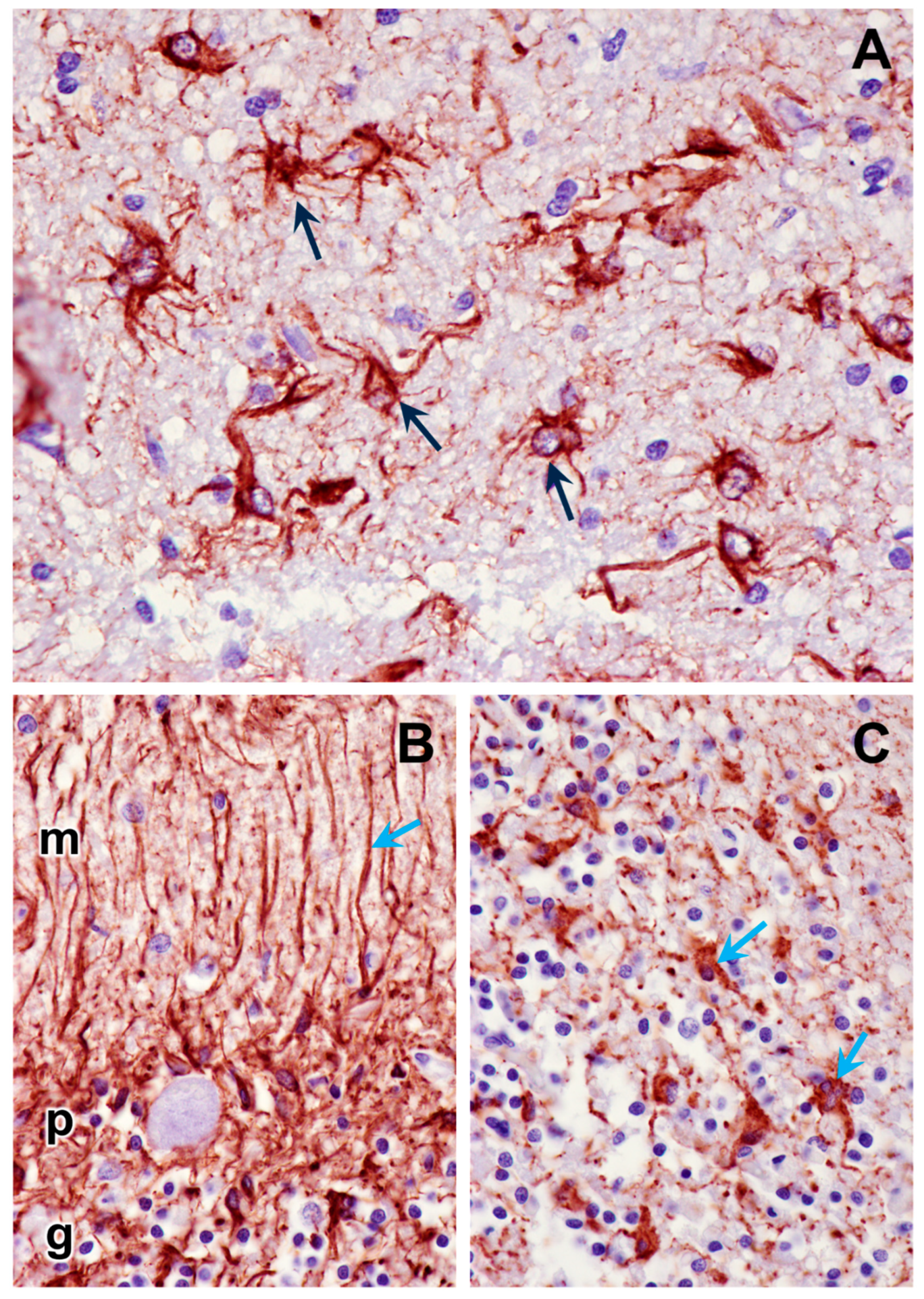
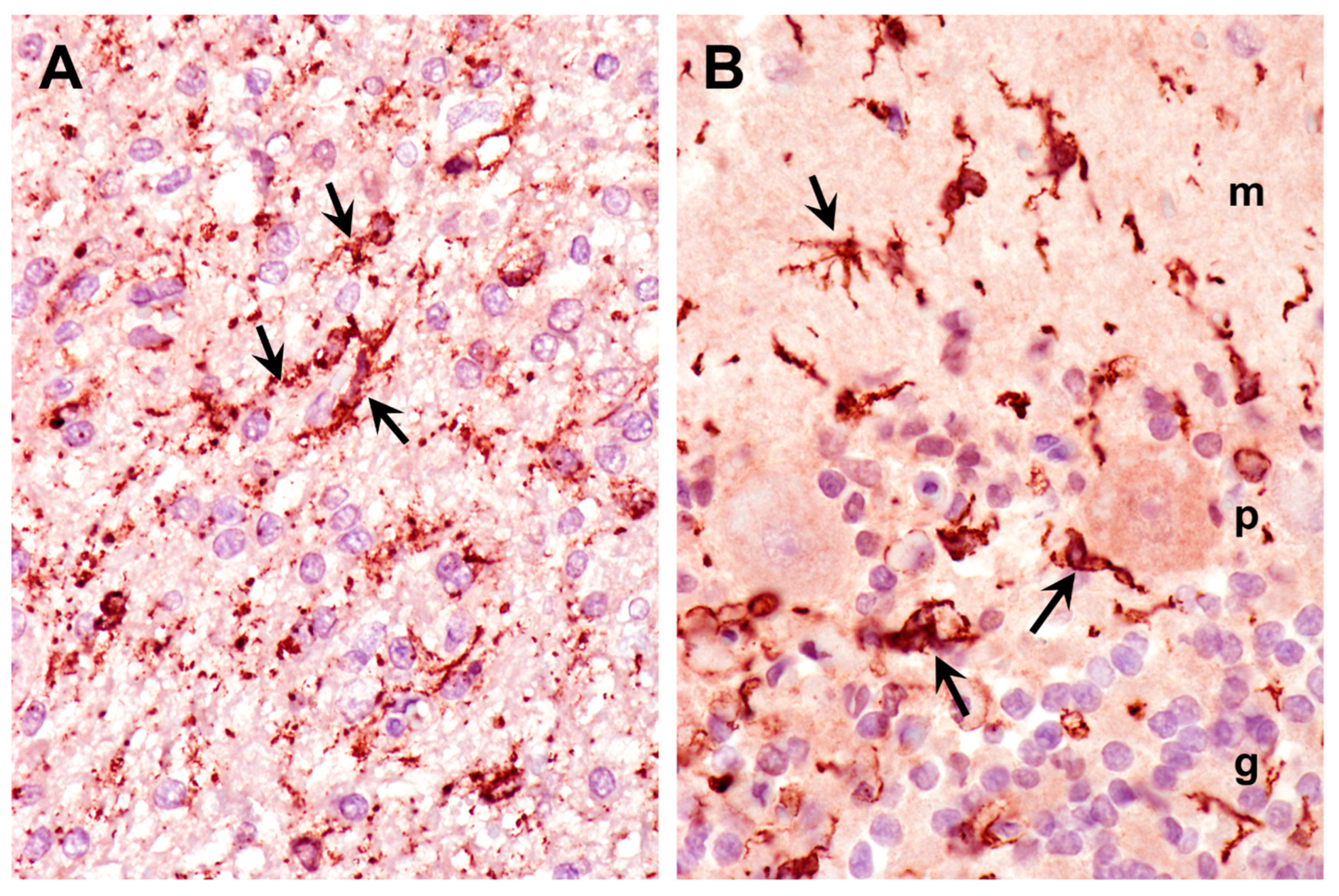
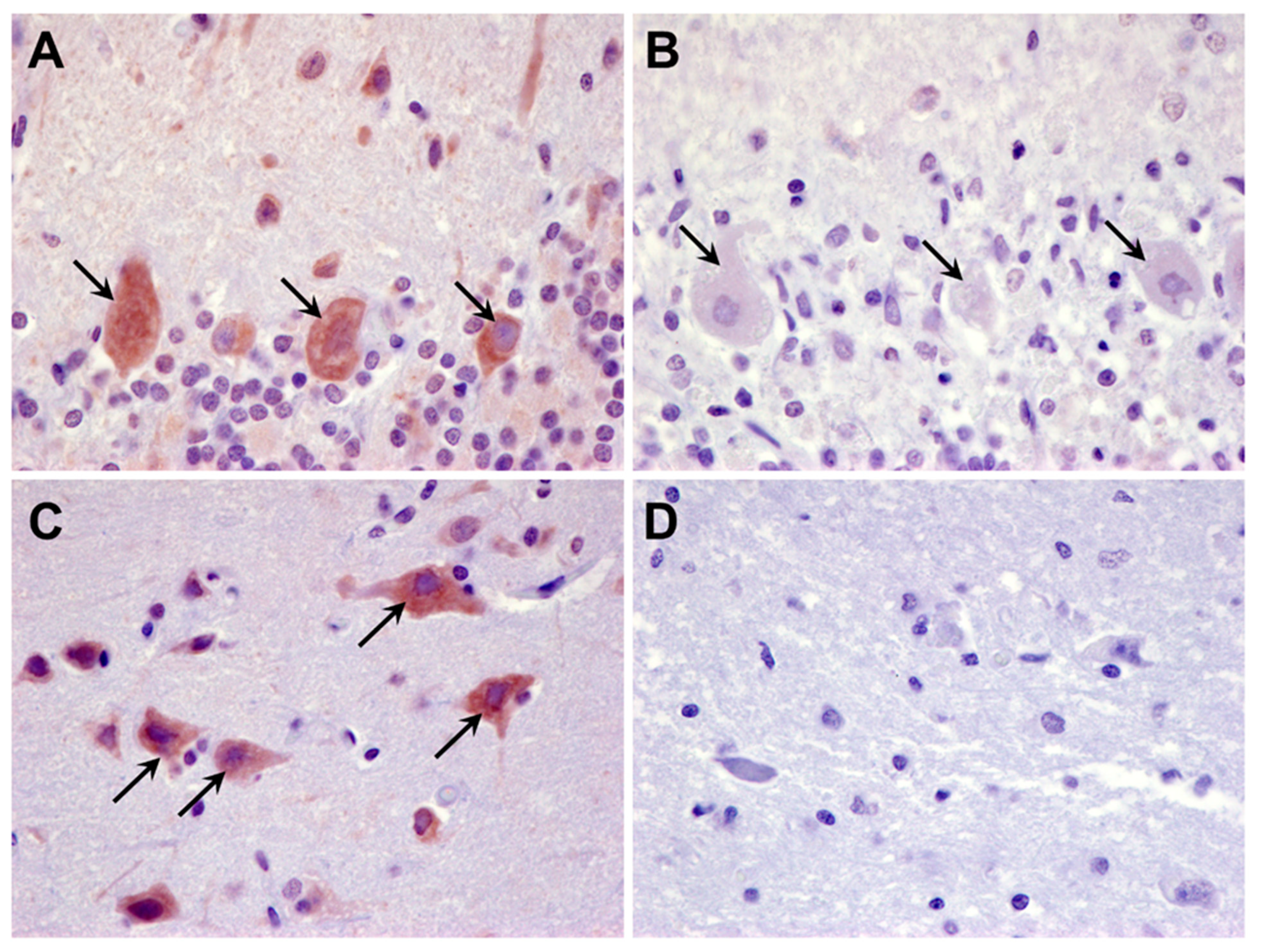
3.1. Microscopic Findings
3.2. Molecular Genetic Findings
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Katz, M.L.; Rustad, E.; Robinson, G.O.; Whiting, R.E.H.; Student, J.T.; Coates, J.R.; Narfstrom, K. Canine Neuronal Ceroid Lipofuscinoses: Promising Models for Preclinical Testing of Therapeutic Interventions. Neurobiol. Dis. 2017, 108, 277–287. [Google Scholar] [CrossRef]
- Butz, E.S.; Chandrachud, U.; Mole, S.E.; Cotman, S.L. Moving towards a New Era of Genomics in the Neuronal Ceroid Lipofuscinoses. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165571. [Google Scholar] [CrossRef]
- Beck-Wodl, S.; Harzer, K.; Sturm, M.; Buchert, R.; Ries, O.; Mennel, H.-D.; Latta, E.; Pagenstecher, A.; Keber, U. Homozygous TBC1 Domain-Containing Kinase (TBCK) Mutation Causes a Novel Lysosomal Storage Disease—A New Type of Neuronal Ceroid Lipofuscinosis (CLN15)? Acta Neuropathol. Commun. 2018, 6, 145. [Google Scholar] [CrossRef]
- Mole, S.E.; Williams, R.E.; Goebel, H.H. The Neuronal Ceroid Lipofuscinoses (Batten Disease), 2nd ed.; Mole, S.E., Willimas, R.E., Goebel, H.H., Eds.; Oxford University Press: Oxford, UK, 2011. [Google Scholar]
- Abitbol, M.; Thibaud, J.-L.; Olby, N.J.; Hitte, C.; Puech, J.-P.; Maurer, M.; Pilot-Storck, F.; Hedan, B.; Dreano, S.; Brahimi, S.; et al. A Canine Arylsulfatase G (ARSG) Mutation Leading to a Sulfatase Deficiency Is Associated with Neuronal Ceroid Lipofuscinosis. Proc. Natl. Acad. Sci. USA 2010, 107, 14775–14780. [Google Scholar] [CrossRef]
- Keller, S.H.; Johnson, G.S.; Bullock, G.; Mhlanga-Mutangadura, T.; Schwartz, M.; Pattridge, S.G.; Guo, J.; Kortz, G.D.; Katz, M.L. Homozygous CNP Mutation and Neurodegeneration in Weimaraners: Myelin Abnormalities and Accumulation of Lipofuscin-like Inclusions. Genes 2024, 15, 246. [Google Scholar] [CrossRef]
- Bullock, G.; Johnson, G.S.; Mhlanga-Mutangadura, T.; Petesch, S.C.; Thompson, S.; Goebbels, S.; Katz, M.L. Lysosomal Storage Disease Associated with a CNP Sequence Variant in Dalmatian Dogs. Gene 2022, 830, 146513. [Google Scholar] [CrossRef]
- Bullock, G.; Johnson, G.S.; Pattridge, S.G.; Mhlanga-Matangadura, T.; Guo, T.; Cook, J.; Campbell, R.S.; Vite, C.H.; Katz, M.L. A Homozygous MAN2B1 Missense Mutation in a Doberman Pinscher Dog with Neurodegeneration, Cytoplasmic Vacuoles, Autofluorescent Storage Granules and an α-Mannosidase Deficiency. Genes 2023, 14, 1746. [Google Scholar] [CrossRef]
- Morgan, B.R.; Coates, J.R.; Johnson, G.C.; Shelton, G.D.; Katz, M.L. Characterization of Thoracic Motor and Sensory Neurons and Spinal Nerve Roots in Canine Degenerative Myelopathy, a Potential Disease Model of Amyotrophic Lateral Sclerosis. J. Neurosci. Res. 2014, 92, 531–541. [Google Scholar] [CrossRef]
- Katz, M.L.; Khan, S.; Awano, T.; Shahid, S.A.; Siakotos, A.N.; Johnson, G.S. A Mutation in the CLN8 Gene in English Setter Dogs with Neuronal Ceroid-Lipofuscinosis. Biochem. Biophys. Res. Commun. 2005, 327, 541–547. [Google Scholar] [CrossRef]
- Bathke, J.; Luhken, G. OVarFlow: A Resource Optimized GATK 4 Based Open Source Variant Calling WorkFlow. BMC Bioinform. 2021, 22, 402. [Google Scholar] [CrossRef]
- Palmer, D.N.; Barry, L.A.; Tyynela, J.; Cooper, J.D. NCL Disease Mechanisms. Biochim. Biophys. Acta 2013, 1832, 1882–1893. [Google Scholar] [CrossRef]
- Katz, M.L.; Farias, F.H.; Sanders, D.N.; Zeng, R.; Khan, S.; Johnson, G.S.; O’Brien, D.P. A Missense Mutation in Canine CLN6 in an Australian Shepherd with Neuronal Ceroid Lipofuscinosis. J. Biomed. Biotechnol. 2011, 2011, 198042. [Google Scholar] [CrossRef]
- Rus, C.-M.; Weissensteiner, T.; Pereira, C.; Susnea, I.; Danquah, B.D.; Morales Torres, G.; Rocha, M.E.; Cozma, C.; Saravanakumar, D.; Mannepalli, S.; et al. Clinical and Genetic Characterization of a Cohort of 97 CLN6 Patients Tested at a Single Center. Orphanet J. Rare Dis. 2022, 17, 179. [Google Scholar] [CrossRef]
- Hulbert, A.J.; Turner, N.; Hinde, J.; Else, P.; Guderley, H. How Might You Compare Mitochondria from Different Tissues and Different Species? J. Comp. Physiol. B 2006, 176, 93–105. [Google Scholar] [CrossRef]
- Sadeesh, E.M.; Singla, N.; Lahamge, M.S.; Kumari, S.; Ampadi, A.N.; Anuj, M. Tissue Heterogeneity of Mitochondrial Activity, Biogenesis and Mitochondrial Protein Gene Expression in Buffalo. Mol. Biol. Rep. 2023, 50, 5255–5266. [Google Scholar] [CrossRef]
- Hansen, F.M.; Kremer, L.S.; Karayel, O.; Bludau, I.; Larsson, N.-G.; Kuhl, I.; Mann, M. Mitochondrial Phosphoproteomes Are Functionally Specialized across Tissues. Life Sci. Alliance 2024, 7, e202302147. [Google Scholar] [CrossRef]
- Kaminiow, K.; Kozak, S.; Paprocka, J. Recent Insight into the Genetic Basis, Clinical Features, and Diagnostic Methods for Neuronal Ceroid Lipofuscinosis. Int. J. Mol. Sci. 2022, 23, 5729. [Google Scholar] [CrossRef]
- Naseri, N.; Sharma, M.; Velinov, M. Autosomal Dominant Neuronal Ceroid Lipofuscinosis: Clinical Features and Molecular Basis. Clin. Genet. 2021, 99, 111–118. [Google Scholar] [CrossRef]
- Melville, S.A.; Wilson, C.L.; Chiang, C.S.; Studdert, V.P.; Lingaas, F.; Wilton, A.N. A Mutation in Canine CLN5 Causes Neuronal Ceroid Lipofuscinosis in Border Collie Dogs. Genomics 2005, 86, 287–294. [Google Scholar] [CrossRef]
- Hirz, M.; Drogemuller, M.; Schanzer, A.; Jagannathan, V.; Dietschi, E.; Goebel, H.H.; Hecht, W.; Laubner, S.; Schmidt, M.J.; Steffen, F.; et al. Neuronal Ceroid Lipofuscinosis (NCL) Is Caused by the Entire Deletion of CLN8 in the Alpenlandische Dachsbracke Dog. Mol. Genet. Metab. 2017, 120, 269–277. [Google Scholar] [CrossRef]
- Lingaas, F.; Guttersrud, O.-A.; Arnet, E.; Espenes, A. Neuronal Ceroid Lipofuscinosis in Salukis Is Caused by a Single Base Pair Insertion in CLN8. Anim. Genet. 2018, 49, 52–58. [Google Scholar] [CrossRef]
- Kolicheski, A.; Barnes Heller, H.L.; Arnold, S.; Schnabel, R.D.; Taylor, J.F.; Knox, C.A.; Mhlanga-Mutangadura, T.; O’Brien, D.P.; Johnson, G.S.; Dreyfus, J.; et al. Homozygous PPT1 Splice Donor Mutation in a Cane Corso Dog with Neuronal Ceroid Lipofuscinosis. J. Vet. Intern. Med. 2017, 31, 149–157. [Google Scholar] [CrossRef]
- Sanders, D.N.; Farias, F.H.; Johnson, G.S.; Chiang, V.; Cook, J.R.; O’Brien, D.P.; Hofmann, S.L.; Lu, J.-Y.; Katz, M.L. A Mutation in Canine PPT1 Causes Early Onset Neuronal Ceroid Lipofuscinosis in a Dachshund. Mol. Genet. Metab. 2010, 100, 349–356. [Google Scholar] [CrossRef]
- Awano, T.; Katz, M.L.; O’Brien, D.P.; Sohar, I.; Lobel, P.; Coates, J.R.; Khan, S.; Johnson, G.C.; Giger, U.; Johnson, G.S. A Frame Shift Mutation in Canine TPP1 (the Ortholog of Human CLN2) in a Juvenile Dachshund with Neuronal Ceroid Lipofuscinosis. Mol. Genet. Metab. 2006, 89, 254–260. [Google Scholar] [CrossRef]
- Villani, N.A.; Bullock, G.; Michaels, J.R.; Yamato, O.; O’Brien, D.P.; Mhlanga-Mutangadura, T.; Johnson, G.S.; Katz, M.L. A Mixed Breed Dog with Neuronal Ceroid Lipofuscinosis Is Homozygous for a CLN5 Nonsense Mutation Previously Identified in Border Collies and Australian Cattle Dogs. Mol. Genet. Metab. 2019, 127, 107–115. [Google Scholar] [CrossRef]
- Schmutz, I.; Jagannathan, V.; Bartenschlager, F.; Stein, V.M.; Gruber, A.D.; Leeb, T.; Katz, M.L. ATP13A2 Missense Variant in Australian Cattle Dogs with Late Onset Neuronal Ceroid Lipofuscinosis. Mol. Genet. Metab. 2019, 127, 95–106. [Google Scholar] [CrossRef]
- Kolicheski, A.; Johnson, G.S.; O’Brien, D.P.; Mhlanga-Mutangadura, T.; Gilliam, D.; Guo, J.; Anderson-Sieg, T.D.; Schnabel, R.D.; Taylor, J.F.; Lebowitz, A.; et al. Australian Cattle Dogs with Neuronal Ceroid Lipofuscinosis Are Homozygous for a CLN5 Nonsense Mutation Previously Identified in Border Collies. J. Vet. Intern. Med. 2016, 30, 1149–1158. [Google Scholar] [CrossRef]
- Gilliam, D.; Kolicheski, A.; Johnson, G.S.; Mhlanga-Mutangadura, T.; Taylor, J.F.; Schnabel, R.D.; Katz, M.L. Golden Retriever Dogs with Neuronal Ceroid Lipofuscinosis Have a Two-Base-Pair Deletion and Frameshift in CLN5. Mol. Genet. Metab. 2015, 115, 101–109. [Google Scholar] [CrossRef]
- Guo, J.; Johnson, G.S.; Brown, H.A.; Provencher, M.L.; da Costa, R.C.; Mhlanga-Mutangadura, T.; Taylor, J.F.; Schnabel, R.D.; O’Brien, D.P.; Katz, M.L. A CLN8 Nonsense Mutation in the Whole Genome Sequence of a Mixed Breed Dog with Neuronal Ceroid Lipofuscinosis and Australian Shepherd Ancestry. Mol. Genet. Metab. 2014, 112, 302–309. [Google Scholar] [CrossRef]
- Ashwini, A.; D’Angelo, A.; Yamato, O.; Giordano, C.; Cagnotti, G.; Harcourt-Brown, T.; Mhlanga-Mutangadura, T.; Guo, J.; Johnson, G.S.; Katz, M.L. Neuronal Ceroid Lipofuscinosis Associated with an MFSD8 Mutation in Chihuahuas. Mol. Genet. Metab. 2016, 118, 326–332. [Google Scholar] [CrossRef]
- Guo, J.; O’Brien, D.P.; Mhlanga-Mutangadura, T.; Olby, N.J.; Taylor, J.F.; Schnabel, R.D.; Katz, M.L.; Johnson, G.S. A Rare Homozygous MFSD8 Single-Base-Pair Deletion and Frameshift in the Whole Genome Sequence of a Chinese Crested Dog with Neuronal Ceroid Lipofuscinosis. BMC Vet. Res. 2015, 10, 960. [Google Scholar] [CrossRef]
- Guo, J.; Johnson, G.S.; Cook, J.; Harris, O.K.; Mhlanga-Mutangadura, T.; Schnabel, R.D.; Jensen, C.A.; Katz, M.L. Neuronal Ceroid Lipofuscinosis in a German Shorthaired Pointer Associated with a Previously Reported CLN8 Nonsense Variant. Mol. Genet. Metab. Rep. 2019, 21, 100521. [Google Scholar] [CrossRef]
- Awano, T.; Katz, M.L.; O’Brien, D.P.; Taylor, J.F.; Evans, J.; Khan, S.; Sohar, I.; Lobel, P.; Johnson, G.S. A Mutation in the Cathepsin D Gene (CTSD) in American Bulldogs with Neuronal Ceroid Lipofuscinosis. Mol. Genet. Metab. 2006, 87, 341–348. [Google Scholar] [CrossRef]
- Farias, F.H.G.; Zeng, R.; Johnson, G.S.; Wininger, F.A.; Taylor, J.F.; Schnabel, R.D.; McKay, S.D.; Sanders, D.N.; Lohi, H.; Seppälä, E.H.; et al. A Truncating Mutation in ATP13A2 Is Responsible for Adult-Onset Neuronal Ceroid Lipofuscinosis in Tibetan Terriers. Neurobiol. Dis. 2011, 42, 468–474. [Google Scholar] [CrossRef]
- Sharp, J.D.; Wheeler, R.B.; Parker, K.A.; Gardiner, R.M.; Williams, R.E.; Mole, S.E. Spectrum of CLN6 Mutations in Variant Late Infantile Neuronal Ceroid Lipofuscinosis. Hum. Mutat. 2003, 22, 35–42. [Google Scholar] [CrossRef]
- Teixeira, C.A.; Espinola, J.; Huo, L.; Kohlschutter, J.; Persaud Sawin, D.-A.; Minassian, B.; Bessa, C.J.P.; Guimaraes, A.; Stephan, D.A.; Sa Miranda, M.C.; et al. Novel Mutations in the CLN6 Gene Causing a Variant Late Infantile Neuronal Ceroid Lipofuscinosis. Hum. Mutat. 2003, 21, 502–508. [Google Scholar] [CrossRef]
- Bouhouche, A.; Regragui, W.; El Fahime, E.; Bouslam, N.; Tazi-Ahnini, R.; Melloul, M.; Benomar, A.; Yahyaoui, M. CLN6 p.I154del Mutation Causing Late Infantile Neuronal Ceroid Lipofuscinosis in a Large Consanguineous Moroccan Family. Indian J. Pediatr. 2013, 80, 694–696. [Google Scholar] [CrossRef]
- Chin, J.J.; Behnam, B.; Davids, M.; Sharma, P.; Zein, W.M.; Wang, C.; Chepa-Lotrea, X.; Gallantine, W.B.; Toro, C.; Adams, D.R.; et al. Novel Mutations in CLN6 Cause Late-Infantile Neuronal Ceroid Lipofuscinosis without Visual Impairment in Two Unrelated Patients. Mol. Genet. Metab. 2019, 126, 188–195. [Google Scholar] [CrossRef]
- Onodera, M.; Tsujimoto, S.; Doi, S.; Yamashita, A.; Yamazaki, T.; Makifuchi, T.; Inazu, T.P. Asn77Lys Homozygous CLN6 Mutation in Two Unrelated Japanese Patients with Kufs Disease, an Adult Onset Neuronal Ceroid Lipofuscinosis. Clin. Chim. Acta 2021, 523, 191–195. [Google Scholar] [CrossRef]
- Sun, G.; Yao, F.; Tian, Z.; Ma, T.; Yang, Z. A First CLN6 Variant Case of Late Infantile Neuronal Ceroid Lipofuscinosis Caused by a Homozygous Mutation in a Boy from China: A Case Report. BMC Med. Genet. 2018, 19, 177. [Google Scholar] [CrossRef]
- Sato, R.; Inui, T.; Endo, W.; Okubo, Y.; Takezawa, Y.; Anzai, M.; Morita, H.; Saitsu, H.; Matsumoto, N.; Haginoya, K. First Japanese Variant of Late Infantile Neuronal Ceroid Lipofuscinosis Caused by Novel CLN6 Mutations. Brain Dev. 2016, 38, 852–856. [Google Scholar] [CrossRef]
- Matsumoto, A.; Nagashima, M.; Iwama, K.; Mizuguchi, T.; Makino, S.; Ikeda, T.; Muramatsu, K.; Matsumoto, N.; Yamagata, T.; Osaka, H. Rapid Progression of a Walking Disability in a 5-Year-Old Boy with a CLN6 Mutation. Brain Dev. 2019, 41, 726–730. [Google Scholar] [CrossRef]
- Panjeshahi, S.; Karimzadeh, P.; Movafagh, A.; Ahmadabadi, F.; Rahimian, E.; Alijanpour, S.; Miryounesi, M. Clinical and Genetic Characterization of Neuronal Ceroid Lipofuscinoses (NCLs) in 29 Iranian Patients: Identification of 11 Novel Mutations. Hum. Genet. 2023, 142, 1001–1016. [Google Scholar] [CrossRef]
- Kousi, M.; Lehesjoki, A.-E.; Mole, S.E. Update of the Mutation Spectrum and Clinical Correlations of over 360 Mutations in Eight Genes That Underlie the Neuronal Ceroid Lipofuscinoses. Hum. Mutat. 2012, 33, 42–63. [Google Scholar] [CrossRef]
- Wheeler, R.B.; Sharp, J.D.; Schultz, R.A.; Joslin, J.M.; Williams, R.E.; Mole, S.E. The Gene Mutated in Variant Late-Infantile Neuronal Ceroid Lipofuscinosis (CLN6) and in Nclf Mutant Mice Encodes a Novel Predicted Transmembrane Protein. Am. J. Hum. Genet. 2002, 70, 537–542. [Google Scholar] [CrossRef]
- Heine, C.; Quitsch, A.; Storch, S.; Martin, Y.; Lonka, L.; Lehesjoki, A.-E.; Mole, S.E.; Braulke, T. Topology and Endoplasmic Reticulum Retention Signals of the Lysosomal Storage Disease-Related Membrane Protein CLN6. Mol. Membr. Biol. 2007, 24, 74–87. [Google Scholar] [CrossRef]
- Mole, S.E.; Michaux, G.; Codlin, S.; Wheeler, R.B.; Sharp, J.D.; Cutler, D.F. CLN6, Which Is Associated with a Lysosomal Storage Disease, Is an Endoplasmic Reticulum Protein. Exp. Cell Res. 2004, 298, 399–406. [Google Scholar] [CrossRef]
- Rus, C.-M.; Polla, D.L.; Di Bucchianico, S.; Fischer, S.; Hartkamp, J.; Hartmann, G.; Alpagu, Y.; Cozma, C.; Zimmermann, R.; Bauer, P. Neuronal Progenitor Cells-Based Metabolomics Study Reveals Dysregulated Lipid Metabolism and Identifies Putative Biomarkers for CLN6 Disease. Sci. Rep. 2023, 13, 18550. [Google Scholar] [CrossRef]
- Tuermer, A.; Mausbach, S.; Kaade, E.; Damme, M.; Sylvester, M.; Gieselmann, V.; Thelen, M. CLN6 Deficiency Causes Selective Changes in the Lysosomal Protein Composition. Proteomics 2021, 21, e2100043. [Google Scholar] [CrossRef]
- Bajaj, L.; Sharma, J.; di Ronza, A.; Zhang, P.; Eblimit, A.; Pal, R.; Roman, D.; Collette, J.R.; Booth, C.; Chang, K.T.; et al. A CLN6-CLN8 Complex Recruits Lysosomal Enzymes at the ER for Golgi Transfer. J. Clin. Investig. 2020, 130, 4118–4132. [Google Scholar] [CrossRef]
- Yamashita, A.; Hiraki, Y.; Yamazaki, T. Identification of CLN6 as a Molecular Entity of Endoplasmic Reticulum-Driven Anti-Aggregate Activity. Biochem. Biophys. Res. Commun. 2017, 487, 917–922. [Google Scholar] [CrossRef]
- Yamashita, A.; Shiro, Y.; Hiraki, Y.; Yujiri, T.; Yamazaki, T. Implications of Graded Reductions in CLN6′s Anti-Aggregate Activity for the Development of the Neuronal Ceroid Lipofuscinoses. Biochem. Biophys. Res. Commun. 2020, 525, 883–888. [Google Scholar] [CrossRef]
- Shiro, Y.; Yamashita, A.; Watanabe, K.; Yamazaki, T. CLN6′s Luminal Tail-Mediated Functional Interference between CLN6 Mutants as a Novel Pathomechanism for the Neuronal Ceroid Lipofuscinoses. Biomed. Res. 2021, 42, 129–138. [Google Scholar] [CrossRef]
- Best, H.L.; Clare, A.J.; McDonald, K.O.; Wicky, H.E.; Hughes, S.M. An Altered Secretome Is an Early Marker of the Pathogenesis of CLN6 Batten Disease. J. Neurochem. 2021, 157, 764–780. [Google Scholar] [CrossRef]
- Teixeira, C.A.F.; Lin, S.; Mangas, M.; Quinta, R.; Bessa, C.J.P.; Ferreira, C.; Sa Miranda, M.C.; Boustany, R.-M.N.; Ribeiro, M.G. Gene Expression Profiling in VLINCL CLN6-Deficient Fibroblasts: Insights into Pathobiology. Biochim. Biophys. Acta 2006, 1762, 637–646. [Google Scholar] [CrossRef]
- Palmer, D.N.; Fearnley, I.M.; Walker, J.E.; Hall, N.A.; Lake, B.D.; Wolfe, L.S.; Haltia, M.; Martinus, R.D.; Jolly, R.D. Mitochondrial ATP Synthase Subunit c Storage in the Ceroid-Lipofuscinoses (Batten Disease). Am. J. Med. Genet. 1992, 42, 561–567. [Google Scholar] [CrossRef]
- Martinus, R.D.; Harper, P.A.; Jolly, R.D.; Bayliss, S.L.; Midwinter, G.G.; Shaw, G.J.; Palmer, D.N. Bovine Ceroid-Lipofuscinosis (Batten’s Disease): The Major Component Stored Is the DCCD-Reactive Proteolipid, Subunit C, of Mitochondrial ATP Synthase. Vet. Res. Commun. 1991, 15, 85–94. [Google Scholar] [CrossRef]
- Palmer, D.N.; Fearnley, I.M.; Medd, S.M.; Walker, J.E.; Martinus, R.D.; Bayliss, S.L.; Hall, N.A.; Lake, B.D.; Wolfe, L.S.; Jolly, R.D. Lysosomal Storage of the DCCD Reactive Proteolipid Subunit of Mitochondrial ATP Synthase in Human and Ovine Ceroid Lipofuscinoses. Adv. Exp. Med. Biol. 1989, 266, 211–223. [Google Scholar] [CrossRef]
- Fearnley, I.M.; Walker, J.E.; Martinus, R.D.; Jolly, R.D.; Kirkland, K.B.; Shaw, G.J.; Palmer, D.N. The Sequence of the Major Protein Stored in Ovine Ceroid Lipofuscinosis Is Identical with That of the Dicyclohexylcarbodiimide-Reactive Proteolipid of Mitochondrial ATP Synthase. Biochem. J. 1990, 268, 751–758. [Google Scholar] [CrossRef]
- Hughes, S.M.; Moroni-Rawson, P.; Jolly, R.D.; Jordan, T.W. Submitochondrial Distribution and Delayed Proteolysis of Subunit c of the H+-Transporting ATP-Synthase in Ovine Ceroid-Lipofuscinosis. Electrophoresis 2001, 22, 1785–1794. [Google Scholar] [CrossRef]
- Kominami, E.; Ezaki, J.; Wolfe, L.S. New Insight into Lysosomal Protein Storage Disease: Delayed Catabolism of ATP Synthase Subunit c in Batten Disease. Neurochem. Res. 1995, 20, 1305–1309. [Google Scholar] [CrossRef]
- Tanner, A.J.; Dice, J.F. Batten Disease and Mitochondrial Pathways of Proteolysis. Biochem. Mol. Med. 1996, 57, 1–9. [Google Scholar] [CrossRef]
- Das, A.M.; Jolly, R.D.; Kohlschutter, A. Anomalies of Mitochondrial ATP Synthase Regulation in Four Different Types of Neuronal Ceroid Lipofuscinosis. Mol. Genet. Metab. 1999, 66, 349–355. [Google Scholar] [CrossRef]
- Pezzini, F.; Gismondi, F.; Tessa, A.; Tonin, P.; Carrozzo, R.; Mole, S.E.; Santorelli, F.M.; Simonati, A. Involvement of the Mitochondrial Compartment in Human NCL Fibroblasts. Biochem. Biophys. Res. Commun. 2011, 416, 159–164. [Google Scholar] [CrossRef]
- Cao, Y.; Staropoli, J.F.; Biswas, S.; Espinola, J.A.; MacDonald, M.E.; Lee, J.-M.; Cotman, S.L. Distinct Early Molecular Responses to Mutations Causing VLINCL and JNCL Presage ATP Synthase Subunit C Accumulation in Cerebellar Cells. PLoS ONE 2011, 6, e17118. [Google Scholar] [CrossRef]
- Heine, C.; Tyynela, J.; Cooper, J.D.; Palmer, D.N.; Elleder, M.; Kohlschutter, A.; Braulke, T. Enhanced Expression of Manganese-Dependent Superoxide Dismutase in Human and Sheep CLN6 Tissues. Biochem. J. 2003, 376, 369–376. [Google Scholar] [CrossRef]
- Nathanson, J.; Swarr, D.T.; Singer, A.; Liu, M.; Chinn, A.; Jones, W.; Hurst, J.; Khalek, N.; Zackai, E.; Slavotinek, A. Novel FREM1 mutations expand the phenotypic spectrum associated with Manitoba-oculo-tricho-anal (MOTA) syndrome and bifid nose renal agenesis anorectal malformations (BNAR) syndrome. Am. J. Med. Genet. 2013, 161A, 473–478. [Google Scholar] [CrossRef]




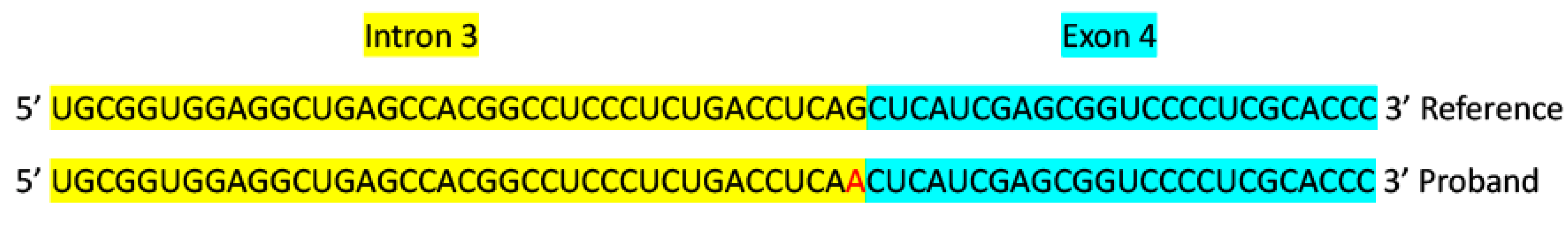

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mhlanga-Mutangadura, T.; Bullock, G.; Cerda-Gonzalez, S.; Katz, M.L. Neuronal Ceroid Lipofuscinosis in a Mixed-Breed Dog with a Splice Site Variant in CLN6. Genes 2024, 15, 661. https://doi.org/10.3390/genes15060661
Mhlanga-Mutangadura T, Bullock G, Cerda-Gonzalez S, Katz ML. Neuronal Ceroid Lipofuscinosis in a Mixed-Breed Dog with a Splice Site Variant in CLN6. Genes. 2024; 15(6):661. https://doi.org/10.3390/genes15060661
Chicago/Turabian StyleMhlanga-Mutangadura, Tendai, Garrett Bullock, Sofia Cerda-Gonzalez, and Martin L. Katz. 2024. "Neuronal Ceroid Lipofuscinosis in a Mixed-Breed Dog with a Splice Site Variant in CLN6" Genes 15, no. 6: 661. https://doi.org/10.3390/genes15060661
APA StyleMhlanga-Mutangadura, T., Bullock, G., Cerda-Gonzalez, S., & Katz, M. L. (2024). Neuronal Ceroid Lipofuscinosis in a Mixed-Breed Dog with a Splice Site Variant in CLN6. Genes, 15(6), 661. https://doi.org/10.3390/genes15060661