
PCR in Forensic Science: A Critical Review



Highlights
- The introduction of PCR has revolutionised forensic DNA profiling.
- With increasing sample complexity and the demands of forensic science, the ability of PCR is being pushed to its limits.
- This paper reviews the different PCR applications and cycling conditions that have been adopted over the years for use in forensic science and suggests where the field may be heading.
Abstract
1. The Polymerase Chain Reaction & Forensic Science
1.1. The General Principles of PCR
1.2. Historial Significance and Applications of PCR in Forensic Science
1.3. Driving Factors of PCR Evolution in Forensic Science
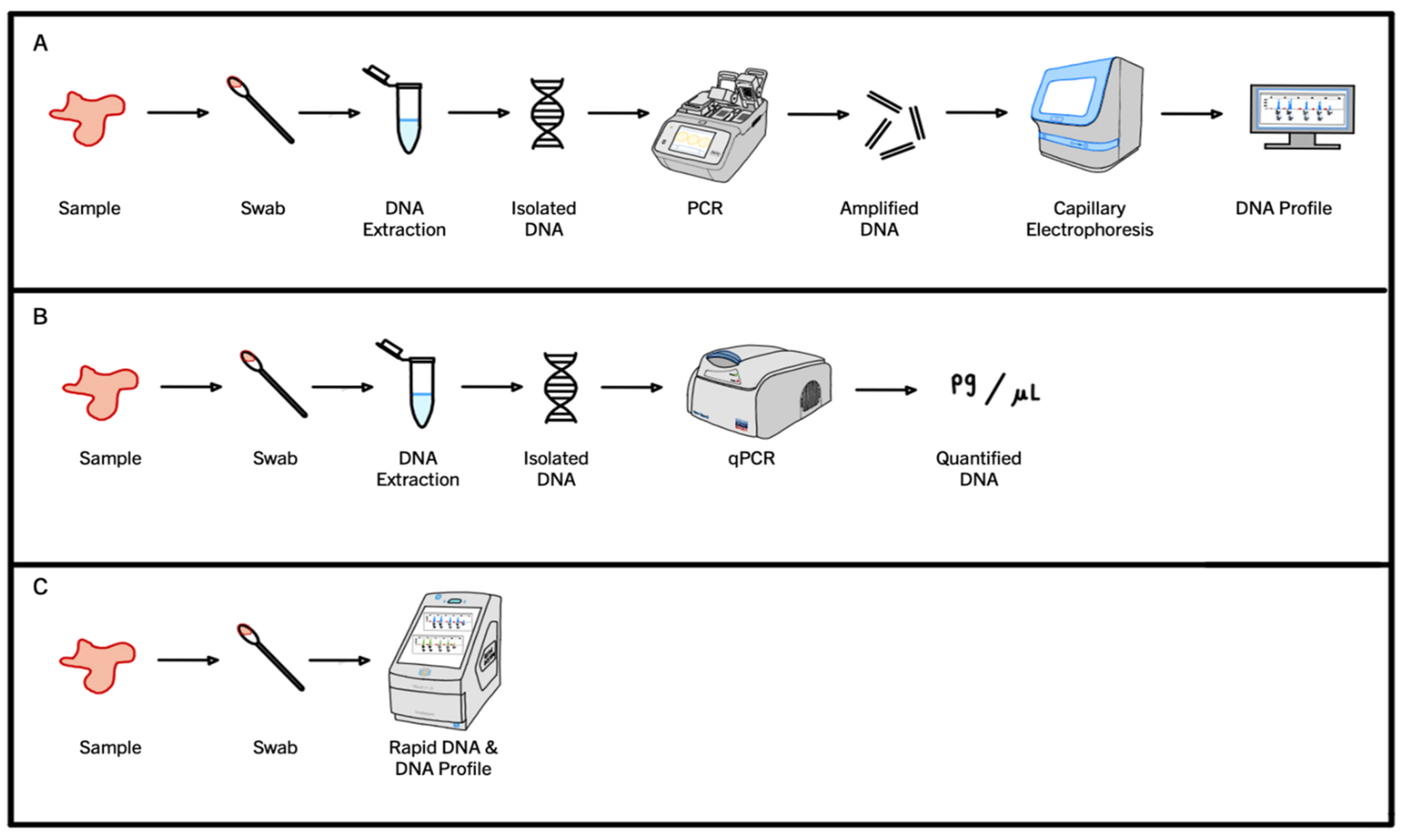
1.4. Current PCR Workflows in Forensic Science
2. Fundamental Factors of the Polymerase Chain Reaction
2.1. PCR Variants—Uniplex and Multiplex PCR
2.2. Factors Influencing PCR Cycling Conditions
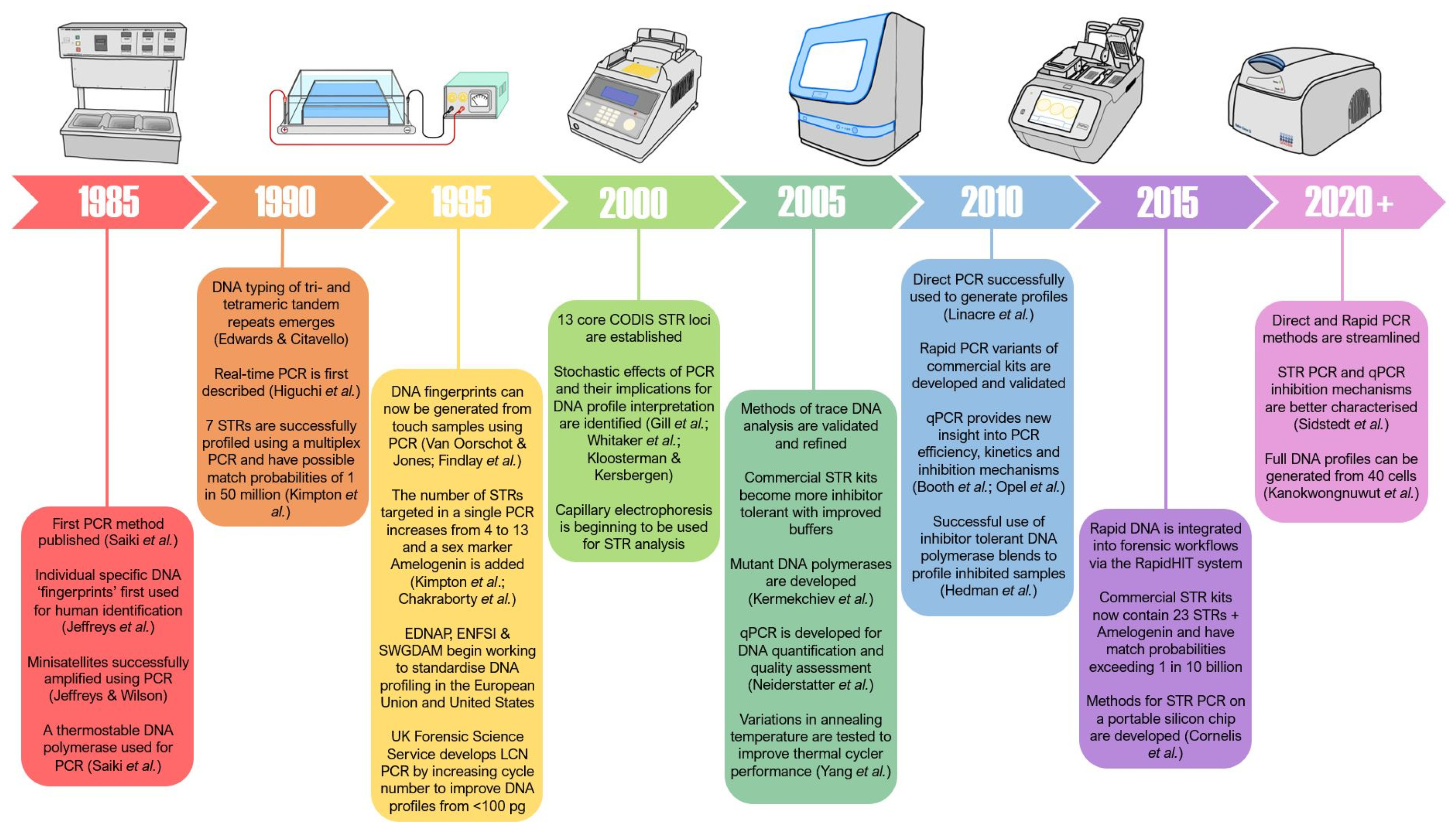
3. Evolution of the Polymerase Chain Reaction
3.1. Evolution of PCR Cycling Conditions in Forensic Science
3.1.1. Short Tandem Repeat Profiling

{kind=link}
{kind=link}
| STR Kit | Type | Year | Cycling Conditions | Total Cycles | Reaction Volume | ||
|---|---|---|---|---|---|---|---|
| Denaturation | Annealing | Extension | |||||
| PowerPlex1.1 [97] and PowerPlex 2.1 [98] | Traditional | 1997 | 94 °C 30 s (10 cycles) 90 °C 30 s (20 cycles) | 60 °C 30 s | 70 °C 45 s | 30 | 22.5 L |
| SGM Plus [99] | Traditional | 1999 | 94 °C 1 min | 59 °C 1 min | 72 °C 1 min | 28 | 50 L |
| PowerPlex 16 [100] | Traditional | 2001 | 94 °C 30 s (10 cycles) 90 °C 30 s (22 cycles) | 60 °C 30 s | 70 °C 45 s | 32 | 25 L |
| AmpFSTR Identifiler [101] | Traditional | 2001 | 94 °C 1 min | 59 °C 1 min | 72 °C 1 min | 28 | 26 L |
| MiniFiler [102] | Traditional | 2007 | 94 °C 20 s | 59 °C 2 min | 72 °C 1 min | 30 | 25 L |
| AmpFSTR Identifiler Plus [103] | Traditional | 2010 | 94 °C 20 s | 59 °C 3 min | 28–29 | 25 L | |
| AmpFSTR NGM Select Express [104] | Rapid | 2011 | 94 °C 3 s | 59 C 16 s | 65 °C 29 s | 25–28 | 25 L |
| PowerPlex 21 [32] | Traditional | 2012 | 94 °C 10 s | 59 °C 1 min | 72 °C 30 s | 30 | 25 L |
| GlobalFiler and GlobalFiler IQC [35] | Traditional | 2013 | 94 °C 10 s | 59 °C 90 s | 29–30 | 25 L | |
| GlobalFiler Express [105] | Rapid | 2015 | 94 °C 3 s | 60 °C 60 s | 25–28 | 15 L | |
| VeriFiler Plus [106] | Traditional | 2018 | 96 °C 10 s | 62 °C 90 s (2 cycles) 59 °C 90 s (27 cycles) | 29 | 25 L | |
| VeriFiler Express [107] | Rapid | 2021 | 96 °C 10 s | 59 C 16 s | 65 °C 29 s | 25–28 | 25 L |
3.1.2. Mitochondrial DNA Testing
3.1.3. Single Nucleotide Polymorphism Analysis
3.2. Evolution of PCR Cycling Conditions in Other Disciplines
4. Recent Developments in PCR for DNA Profiling
4.1. Increased Speed
4.2. Increased Sensitivity and Discrimination Power
4.3. Optimization of Commercially Available Kits
4.4. PCR Amplification Kinetics
5. Recent Developments in PCR beyond Forensics
6. Remaining Challenges in PCR for DNA Profiling
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lorenz, T.C. Polymerase chain reaction: Basic protocol plus troubleshooting and optimization strategies. J. Vis. Exp. 2012, 63, e3998. [Google Scholar] [CrossRef]
- Microbiology, B.O. The polymerase chain reaction: An overview and development of diagnostic PCR protocols at the LCDC. Can. J. Infect. Dis. 1991, 2, 89–91. [Google Scholar] [CrossRef]
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Erlich, H. Specific enzymatic amplification of DNA in vitro: The polymerase chain reaction. Cold Spring Harb. Symp. Quant. Biol. 1986, 51 Pt 1, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Jeffreys, A.J.; Wilson, V.; Neumann, R.; Keyte, J. Amplification of human minisatellites by the polymerase chain reaction: Towards DNA fingerprinting of single cells. Nucleic Acids Res. 1988, 16, 10953–10971. [Google Scholar] [CrossRef] [PubMed]
- Saiki, R.K.; Gelfand, D.H.; Stoffel, S.; Scharf, S.J.; Higuchi, R.; Horn, G.T.; Mullis, K.B.; Erlich, H.A. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 1988, 239, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Holland, P.M.; Abramson, R.D.; Watson, R.; Gelfand, D.H. Detection of specific polymerase chain reaction product by utilizing the 5’----3’ exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. USA 1991, 88, 7276–7280. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.-J.; Kloss-Brandstätter, A.; Richards, M.B.; Yao, Y.-G.; Logan, I. The case for the continuing use of the revised Cambridge Reference Sequence (rCRS) and the standardization of notation in human mitochondrial DNA studies. J. Hum. Genet. 2014, 59, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.; Jeffreys, A.J.; Werrett, D.J. Forensic application of DNA ‘fingerprints’. Nature 1985, 318, 577–579. [Google Scholar] [CrossRef]
- Comey, C.T.; Budowle, B.; Adams, D.E.; Baumstark, A.L.; Lindsey, J.A.; Presley, L.A. PCR amplification and typing of the HLA DQ α gene in forensic samples. J. Forensic Sci. 1993, 38, 239–249. [Google Scholar] [CrossRef]
- Harrington, C.S.; Dunaiski, V.; Williams, K.E.; Fowler, C. HLA DQ α typing of forensic specimens by amplification restriction fragment polymorphism (ARFP) analysis. Forensic Sci. Int. 1991, 51, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, A.D.; Budowle, B.; Daselaar, P. PCR-amplification and detection of the human D1S80 VNTR locus. Amplification conditions, population genetics and application in forensic analysis. Int. J. Leg. Med. 1993, 105, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Sajantila, A.; Budowle, B.; Ström, M.; Johnsson, V.; Lukka, M.; Peltonen, L.; Ehnholm, C. PCR amplification of alleles at the DIS80 locus: Comparison of a Finnish and a North American Caucasian population sample, and forensic casework evaluation. Am. J. Hum. Genet. 1992, 50, 816–825. [Google Scholar] [PubMed]
- Jeffreys, A.J.; Wilson, V.; Thein, S.L. Individual-specific ‘fingerprints’ of human DNA. Nature 1985, 316, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Erlich, H. Part 1.1 in the Beginning: Forensic Applications of DNA Technologies in Silent Witness: Forensic DNA Evidence in Criminal Investigations and Humanitarian Disasters; Oxford University Press: Oxford, UK, 2020. [Google Scholar]
- Stoneking, M.; Hedgecock, D.; Higuchi, R.G.; Vigilant, L.; Erlich, H.A. Population variation of human mtDNA control region sequences detected by enzymatic amplification and sequence-specific oligonucleotide probes. Am. J. Hum. Genet. 1991, 48, 370–382. [Google Scholar] [PubMed]
- Amorim, A.; Fernandes, T.; Taveira, N. Mitochondrial DNA in human identification: A review. PeerJ 2019, 7, e7314. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.; Ivanov, P.L.; Kimpton, C.; Piercy, R.; Benson, N.; Tully, G.; Evett, I.; Hagelberg, E.; Sullivan, K. Identification of the remains of the Romanov family by DNA analysis. Nat. Genet. 1994, 6, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Kimpton, C.; Fisher, D.; Watson, S.; Adams, M.; Urquhart, A.; Lygo, J.; Gill, P. Evaluation of an automated DNA profiling system employing multiplex amplification of four tetrameric STR loci. Int. J. Leg. Med. 1994, 106, 302–311. [Google Scholar] [CrossRef]
- Butler, J.M. The future of forensic DNA analysis. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140252. [Google Scholar] [CrossRef]
- Werrett, D.J. The National DNA Database. Forensic Sci. Int. 1997, 88, 33–42. [Google Scholar] [CrossRef]
- Martin, P.D.; Schmitter, H.; Schneider, P.M. A brief history of the formation of DNA databases in forensic science within Europe. Forensic Sci. Int. 2001, 119, 225–231. [Google Scholar] [CrossRef]
- Butler, J.M. Advanced Topics in Forensic DNA Typing: Methodology; Elsevier Science: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Panneerchelvam, S.; Norazmi, M.N. Forensic DNA profiling and database. Malays. J. Med. Sci. 2003, 10, 20–26. [Google Scholar] [PubMed]
- Jakovski, Z.; Ajanovska, R.J.; Stankov, A.; Poposka, V.; Bitoljanu, N.; Belakaposka, V. The power of forensic DNA data bases in solving crime cases. Forensic Sci. Int. Genet. Suppl. Ser. 2017, 6, e275–e276. [Google Scholar] [CrossRef]
- Ge, J.; Eisenberg, A.; Budowle, B. Developing criteria and data to determine best options for expanding the core CODIS loci. Investig. Genet. 2012, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.; Fereday, L.; Morling, N.; Schneider, P.M. The evolution of DNA databases—Recommendations for new European STR loci. Forensic Sci. Int. 2006, 156, 242–244. [Google Scholar] [CrossRef]
- Margarita, G.; María Victoria, L.; Carmela, P.; Antonio, S.; Angel, C. Ethical-legal problems of DNA databases in criminal investigation. J. Med. Ethics 2000, 26, 266. [Google Scholar] [CrossRef]
- Williams, R.; Johnson, P. Inclusiveness, effectiveness and intrusiveness: Issues in the developing uses of DNA profiling in support of criminal investigations. J. Law. Med. Ethics 2005, 33, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Wallace, H.M.; Jackson, A.R.; Gruber, J.; Thibedeau, A.D. Forensic DNA databases–Ethical and legal standards: A global review. Egypt. J. Forensic Sci. 2014, 4, 57–63. [Google Scholar] [CrossRef]
- Butler, J.M. Recent advances in forensic biology and forensic DNA typing: INTERPOL review 2019–2022. Forensic Sci. Int. Synerg. 2023, 6, 100311. [Google Scholar] [CrossRef]
- Corporation, P. PowerPlex® 21 System for Use on the Applied Biosystems® Genetic Analyzers. Available online: https://www.promega.com/-/media/files/resources/protocols/technical-manuals/tmd/powerplex-21-system-protocol.pdf?rev=7db853167600419eb6ddb19c0e88d4ab&sc_lang=en (accessed on 13 April 2023).
- Ludeman, M.J.; Zhong, C.; Mulero, J.J.; Lagacé, R.E.; Hennessy, L.K.; Short, M.L.; Wang, D.Y. Developmental validation of GlobalFiler™ PCR amplification kit: A 6-dye multiplex assay designed for amplification of casework samples. Int. J. Leg. Med. 2018, 132, 1555–1573. [Google Scholar] [CrossRef]
- Technologies, L. AmpFlSTR® Identifiler® Direct PCR Amplification Kit User Guide. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/cms_065522.pdf (accessed on 22 May 2023).
- Technologies, L. GlobalFiler™ and GlobalFiler™ IQC PCR Amplification Kits: User Guide. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/4477604.pdf (accessed on 22 May 2023).
- Gill, P. Application of low copy number DNA profiling. Croat. Med. J. 2001, 42, 229–232. [Google Scholar] [PubMed]
- Bonsu, D.O.M.; Higgins, D.; Austin, J.J. Forensic touch DNA recovery from metal surfaces—A review. Sci. Justice 2020, 60, 206–215. [Google Scholar] [CrossRef]
- Burrill, J.; Daniel, B.; Frascione, N. A review of trace “Touch DNA” deposits: Variability factors and an exploration of cellular composition. Forensic Sci. Int. Genet. 2019, 39, 8–18. [Google Scholar] [CrossRef]
- Tozzo, P.; Mazzobel, E.; Marcante, B.; Delicati, A.; Caenazzo, L. Touch DNA Sampling Methods: Efficacy Evaluation and Systematic Review. Int. J. Mol. Sci. 2022, 23, 15541. [Google Scholar] [CrossRef]
- van Oorschot, R.A.; Ballantyne, K.N.; Mitchell, R.J. Forensic trace DNA: A review. Investig. Genet. 2010, 1, 14. [Google Scholar] [CrossRef]
- Cook, R.; Mitchell, N.; Henry, J. Assessment of Diamond™ Nucleic Acid Dye for the identification and targeted sampling of latent DNA in operational casework. Forensic Sci. Int. Genet. 2021, 55, 102579. [Google Scholar] [CrossRef] [PubMed]
- Dziak, R.; Peneder, A.; Buetter, A.; Hageman, C. Trace DNA Sampling Success from Evidence Items Commonly Encountered in Forensic Casework. J. Forensic Sci. 2018, 63, 835–841. [Google Scholar] [CrossRef]
- Castella, V.; Mangin, P. DNA profiling success and relevance of 1739 contact stains from caseworks. Forensic Sci. Int. Genet. Suppl. Ser. 2008, 1, 405–407. [Google Scholar] [CrossRef]
- Wong, H.Y.; Tan, J.; Lim, Z.G.; Kwok, R.; Lim, W.; Syn, C.K.-C. DNA profiling success rates of commonly submitted crime scene items. Forensic Sci. Int. Genet. Suppl. Ser. 2019, 7, 597–599. [Google Scholar] [CrossRef]
- Mapes, A.A.; Kloosterman, A.D.; de Poot, C.J. DNA in the Criminal Justice System: The DNA Success Story in Perspective. J. Forensic Sci. 2015, 60, 851–856. [Google Scholar] [CrossRef]
- Mapes, A.A.; Kloosterman, A.D.; van Marion, V.; de Poot, C.J. Knowledge on DNA Success Rates to Optimize the DNA Analysis Process: From Crime Scene to Laboratory. J. Forensic Sci. 2016, 61, 1055–1061. [Google Scholar] [CrossRef]
- Raymond, J.J.; van Oorschot, R.A.H.; Gunn, P.R.; Walsh, S.J.; Roux, C. Trace DNA success rates relating to volume crime offences. Forensic Sci. Int. Genet. Suppl. Ser. 2009, 2, 136–137. [Google Scholar] [CrossRef]
- Raymond, J.J.; Walsh, S.J.; Van Oorschot, R.A.; Gunn, P.R.; Roux, C. Trace DNA: An Underutilized Resource or Pandora’s Box? A Review of the use of Trace DNA Analysis in the Investigation of Volume Crime. J. Forensic Identif. 2004, 54, 668–686. [Google Scholar]
- Saiki, R.K.; Scharf, S.; Faloona, F.; Mullis, K.B.; Horn, G.T.; Erlich, H.A.; Arnheim, N. Enzymatic amplification of β-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 1985, 230, 1350–1354. [Google Scholar] [CrossRef]
- Edwards, A.; Civitello, A.; Hammond, H.A.; Caskey, C.T. DNA typing and genetic mapping with trimeric and tetrameric tandem repeats. Am. J. Hum. Genet. 1991, 49, 746–756. [Google Scholar] [PubMed]
- Higuchi, R.; Fockler, C.; Dollinger, G.; Watson, R. Kinetic PCR Analysis: Real-time Monitoring of DNA Amplification Reactions. Bio/Technology 1993, 11, 1026–1030. [Google Scholar] [CrossRef]
- van Oorschot, R.A.H.; Jones, M.K. DNA fingerprints from fingerprints. Nature 1997, 387, 767. [Google Scholar] [CrossRef]
- Findlay, I.; Taylor, A.; Quirke, P.; Frazier, R.; Urquhart, A. DNA fingerprinting from single cells. Nature 1997, 389, 555–556. [Google Scholar] [CrossRef]
- Kimpton, C.P.; Oldroyd, N.J.; Watson, S.K.; Frazier, R.R.; Johnson, P.E.; Millican, E.S.; Urquhart, A.; Sparkes, B.L.; Gill, P. Validation of highly discriminating multiplex short tandem repeat amplification systems for individual identification. Electrophoresis 1996, 17, 1283–1293. [Google Scholar] [CrossRef]
- Chakraborty, R.; Stivers, D.N.; Su, B.; Zhong, Y.; Budowle, B. The utility of short tandem repeat loci beyond human identification: Implications for development of new DNA typing systems. Electrophoresis 1999, 20, 1682–1696. [Google Scholar] [CrossRef]
- Linacre, A. Review of low template DNA typing. Forensic Sci. Int. Genet. Suppl. Ser. 2009, 2, 549–550. [Google Scholar] [CrossRef]
- Gill, P.; Whitaker, J.; Flaxman, C.; Brown, N.; Buckleton, J. An investigation of the rigor of interpretation rules for STRs derived from less than 100 pg of DNA. Forensic Sci. Int. 2000, 112, 17–40. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, J.P.; Cotton, E.A.; Gill, P. A comparison of the characteristics of profiles produced with the AMPFlSTR® SGM Plus™ multiplex system for both standard and low copy number (LCN) STR DNA analysis. Forensic Sci. Int. 2001, 123, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Kloosterman, A.D.; Kersbergen, P. Efficacy and limits of genotyping low copy number (LCN) DNA samples by multiplex PCR of STR loci. J. Soc. Biol. 2003, 197, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Kermekchiev, M.B.; Kirilova, L.I.; Vail, E.E.; Barnes, W.M. Mutants of Taq DNA polymerase resistant to PCR inhibitors allow DNA amplification from whole blood and crude soil samples. Nucleic Acids Res. 2009, 37, e40. [Google Scholar] [CrossRef]
- Niederstätter, H.; Köchl, S.; Grubwieser, P.; Pavlic, M.; Steinlechner, M.; Parson, W. A modular real-time PCR concept for determining the quantity and quality of human nuclear and mitochondrial DNA. Forensic Sci. Int. Genet. 2007, 1, 29–34. [Google Scholar] [CrossRef]
- Yang, I.; Kim, Y.-H.; Byun, J.-Y.; Park, S.-R. Use of multiplex polymerase chain reactions to indicate the accuracy of the annealing temperature of thermal cycling. Anal. Biochem. 2005, 338, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Linacre, A.; Pekarek, V.; Swaran, Y.C.; Tobe, S.S. Generation of DNA profiles from fabrics without DNA extraction. Forensic Sci. Int. Genet. 2010, 4, 137–141. [Google Scholar] [CrossRef]
- Booth, C.S.; Pienaar, E.; Termaat, J.R.; Whitney, S.E.; Louw, T.M.; Viljoen, H.J. Efficiency of the Polymerase Chain Reaction. Chem. Eng. Sci. 2010, 65, 4996–5006. [Google Scholar] [CrossRef]
- Opel, K.L.; Chung, D.; McCord, B.R. A study of PCR inhibition mechanisms using real time PCR. J. Forensic Sci. 2010, 55, 25–33. [Google Scholar] [CrossRef]
- Hedman, J.; Dufva, C.; Norén, L.; Ansell, C.; Albinsson, L.; Ansell, R. Applying a PCR inhibitor tolerant DNA polymerase blend in forensic DNA profiling. Forensic Sci. Int. Genet. Suppl. Series 2011, 3, e349–e350. [Google Scholar] [CrossRef]
- Cornelis, S.; Fauvart, M.; Gansemans, Y.; Vander Plaetsen, A.-S.; Colle, F.; Wiederkehr, R.S.; Deforce, D.; Stakenborg, T.; Van Nieuwerburgh, F. Multiplex STR amplification sensitivity in a silicon microchip. Sci. Rep. 2018, 8, 9853. [Google Scholar] [CrossRef] [PubMed]
- Sidstedt, M.; Rådström, P.; Hedman, J. PCR inhibition in qPCR, dPCR and MPS-mechanisms and solutions. Anal. Bioanal. Chem. 2020, 412, 2009–2023. [Google Scholar] [CrossRef] [PubMed]
- Kanokwongnuwut, P.; Martin, B.; Taylor, D.; Kirkbride, P.; Linacre, A. How many cells are required for successful DNA profiling? Forensic Sci. Int. Genet. 2021, 51, 102453. [Google Scholar] [CrossRef] [PubMed]
- Moretti, T.R.; Baumstark, A.L.; Defenbaugh, D.A.; Keys, K.M.; Smerick, J.B.; Budowle, B. Validation of short tandem repeats (STRs) for forensic usage: Performance testing of fluorescent multiplex STR systems and analysis of authentic and simulated forensic samples. J. Forensic Sci. 2001, 46, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Chang, C.-W.; Lagacé, R.E.; Calandro, L.M.; Hennessy, L.K. Developmental Validation of the AmpFℓSTR® Identifiler® Plus PCR Amplification Kit: An Established Multiplex Assay with Improved Performance. J. Forensic Sci. 2012, 57, 453–465. [Google Scholar] [CrossRef]
- Ensenberger, M.G.; Hill, C.R.; McLaren, R.S.; Sprecher, C.J.; Storts, D.R. Developmental validation of the PowerPlex(®) 21 System. Forensic Sci. Int. Genet. 2014, 9, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, L.K.; Mehendale, N.; Chear, K.; Jovanovich, S.; Williams, S.; Park, C.; Gangano, S. Developmental validation of the GlobalFiler® express kit, a 24-marker STR assay, on the RapidHIT® System. Forensic Sci. Int. Genet. 2014, 13, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Y.; Gopinath, S.; Lagacé, R.E.; Norona, W.; Hennessy, L.K.; Short, M.L.; Mulero, J.J. Developmental validation of the GlobalFiler® Express PCR Amplification Kit: A 6-dye multiplex assay for the direct amplification of reference samples. Forensic Sci. Int. Genet. 2015, 19, 148–155. [Google Scholar] [CrossRef]
- Vraneš, M.; Scherer, M.; Elliott, K. Development and validation of the Investigator® Quantiplex Pro Kit for qPCR-based examination of the quantity and quality of human DNA in forensic samples. Forensic Sci. Int. Genet. Suppl. Ser. 2017, 6, e518–e519. [Google Scholar] [CrossRef]
- Holt, A.; Wootton, S.C.; Mulero, J.J.; Brzoska, P.M.; Langit, E.; Green, R.L. Developmental validation of the Quantifiler(®) HP and Trio Kits for human DNA quantification in forensic samples. Forensic Sci. Int. Genet. 2016, 21, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Jothikumar, P.; Hill, V.; Narayanan, J. Design of FRET-TaqMan probes for multiplex real-time PCR using an internal positive control. Biotechniques 2009, 46, 519–524. [Google Scholar] [CrossRef]
- Didenko, V.V. DNA probes using fluorescence resonance energy transfer (FRET): Designs and applications. Biotechniques 2001, 31, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Foster, A.; Laurin, N. Development of a fast PCR protocol enabling rapid generation of AmpFℓSTR® Identifiler® profiles for genotyping of human DNA. Investig. Genet. 2012, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.-H. Chapter 9—Amplification of Nucleic Acids. In Diagnostic Molecular Biology; Shen, C.-H., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 215–247. [Google Scholar]
- Hao, L.; Xie, J.; Chen, S.; Wang, S.; Gong, Z.; Ling, K.S.; Guo, L.; Fan, Z.; Zhou, T. A multiple RT-PCR assay for simultaneous detection and differentiation of latent viruses and apscarviroids in apple trees. J. Virol. Methods 2016, 234, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.; Dorneles, E.M.S.; Diniz, C.; Abreu, V.; Sousa, C.; Alves, J.; Carneiro, A.; Bagano, P.; Spier, S.; Barh, D.; et al. Quadruplex PCR assay for identification of Corynebacterium pseudotuberculosis differentiating biovar Ovis and Equi. BMC Vet. Res. 2017, 13, 290. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Zhang, X.; Gao, Y.; Song, S.; Xu, D.; Yan, L. Development and application of multiplex PCR method for simultaneous detection of seven viruses in ducks. BMC Vet. Res. 2019, 15, 103. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, J.; Jurisic, V.; Tosic, N.; Mrdjanovic, J.; Perin, B.; Pavlovic, S.; Djordjevic, N. Optimization of PCR conditions for amplification of GC-Rich EGFR promoter sequence. J. Clin. Lab. Anal. 2013, 27, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Mann, T.; Humbert, R.; Dorschner, M.; Stamatoyannopoulos, J.; Noble, W.S. A thermodynamic approach to PCR primer design. Nucleic Acids Res. 2009, 37, e95. [Google Scholar] [CrossRef]
- Vallone, P.M.; Butler, J.M. AutoDimer: A screening tool for primer-dimer and hairpin structures. Biotechniques 2004, 37, 226–231. [Google Scholar] [CrossRef]
- Holleley, C.E.; Geerts, P.G. Multiplex Manager 1.0: A cross-platform computer program that plans and optimizes multiplex PCR. Biotechniques 2009, 46, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.M.; Devaney, J.M.; Marino, M.A.; Vallone, P.M. Quality control of PCR primers used in multiplex STR amplification reactions. Forensic Sci. Int. 2001, 119, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.G.; Wang, M.X.; Song, P.; Mao, S.; Wang, Y.; Yang, Y.; Luo, J.; Ren, S.; Zhang, D.Y. Designing highly multiplex PCR primer sets with Simulated Annealing Design using Dimer Likelihood Estimation (SADDLE). Nat. Commun. 2022, 13, 1881. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Z.; Ai, Y. Processivity factor of DNA polymerase and its expanding role in normal and translesion DNA synthesis. Biochim. Biophys. Acta 2010, 1804, 1081–1093. [Google Scholar] [CrossRef] [PubMed]
- Wages, J.M., Jr. Polymerase Chain Reaction. Encycl. Anal. Sci. (Second. Ed.) 2005, 243–250. [Google Scholar] [CrossRef]
- Karantzeni, I.; Ruiz, C.; Liu, C.C.; Licata, V.J. Comparative thermal denaturation of Thermus aquaticus and Escherichia coli type 1 DNA polymerases. Biochem. J. 2003, 374, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Hopwood, A.J.; Hurth, C.; Yang, J.; Cai, Z.; Moran, N.; Lee-Edghill, J.G.; Nordquist, A.; Lenigk, R.; Estes, M.D.; Haley, J.P.; et al. Integrated Microfluidic System for Rapid Forensic DNA Analysis: Sample Collection to DNA Profile. Anal. Chem. 2010, 82, 6991–6999. [Google Scholar] [CrossRef]
- LaRue, B.L.; Moore, A.; King, J.L.; Marshall, P.L.; Budowle, B. An evaluation of the RapidHIT(®) system for reliably genotyping reference samples. Forensic Sci. Int. Genet. 2014, 13, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Ward, D.; Henry, J.; Taylor, D. Analysis of mixed DNA profiles from the RapidHIT™ ID platform using probabilistic genotyping software STRmix™. Forensic Sci. Int. Genet. 2022, 58, 102664. [Google Scholar] [CrossRef]
- Hennessy, L.K.; Franklin, H.; Li, Y.; Buscaino, J.; Chear, K.; Gass, J.; Mehendale, N.; Williams, S.; Jovanovich, S.; Harris, D.; et al. Developmental validation studies on the RapidHIT™ Human DNA Identification System. Forensic Sci. Int. Genet. Suppl. Ser. 2013, 4, e7–e8. [Google Scholar] [CrossRef]
- Greenspoon, S.A.; Lytle, P.J.; Turek, S.A.; Rolands, J.M.; Scarpetta, M.A.; Carr, C.D. Validation of the PowerPlex 1.1 loci for use in human identification. J. Forensic Sci. 2000, 45, 677–683. [Google Scholar] [CrossRef]
- Corporation, P. PowerPlex® 2.1 System Technical Manual. Available online: https://www.promega.com/-/media/files/resources/profiles-in-dna/302/the-geneprint-powerplex-system-for-the-fbi-selection-of-thirteen-codis-core-str-loci.pdf?la=en (accessed on 2 June 2023).
- ThermoFischer Scientific. AmpFlSTR® SGM Plus® PCR Amplification Kit. Available online: https://tools.thermofisher.com/content/sfs/manuals/cms_041049.pdf (accessed on 2 June 2023).
- Corporation, P. PowerPlex® 16 System. Available online: https://www.promega.com/-/media/files/resources/protocols/technical-manuals/tmd/powerplex-16-system-protocol.pdf?rev=390c9836b26044b19ce1f709fc245352&sc_lang=en (accessed on 2 June 2023).
- ThermoFischer Scientific. AmpFlSTR® Identifiler® PCR Amplification Kit. Available online: https://tools.thermofisher.com/content/sfs/manuals/cms_041201.pdf (accessed on 2 June 2023).
- Technologies, L. AmpFlSTR™ MiniFiler™ PCR Amplification Kit User Guide. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/cms_042748.pdf (accessed on 2 June 2023).
- ThermoFischer Scientific. AmpFlSTR® Identifiler® Plus PCR Amplification Kit User Guide. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/4440211_AmpFlSTR_IdentifilerPlus_UG.pdf (accessed on 22 May 2023).
- Technologies, L. AmpFlSTR® NGM SElect™ Express PCR Amplification Kit User Guide. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/cms_104061.pdf (accessed on 2 June 2023).
- Technologies, L. GlobalFiler™ Express PCR Amplification Kit User Guide. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/4477672_GlobalFilerExpress_UG.pdf (accessed on 22 May 2023).
- ThermoFischer Scientific. VeriFiler™ Plus PCR Amplification Kit. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0017493_VeriFilerPlusPCRAmpKit_UG.pdf (accessed on 2 June 2023).
- Technologies, L. VeriFiler™ Express PCR Amplification Kit User Guide. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/100043588_VFE_UG.pdf (accessed on 2 June 2023).
- Greenberg, B.D.; Newbold, J.E.; Sugino, A. Intraspecific nucleotide sequence variability surrounding the origin of replication in human mitochondrial DNA. Gene 1983, 21, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Lutz, S.; Wittig, H.; Weisser, H.J.; Heizmann, J.; Junge, A.; Dimo-Simonin, N.; Parson, W.; Edelmann, J.; Anslinger, K.; Jung, S.; et al. Is it possible to differentiate mtDNA by means of HVIII in samples that cannot be distinguished by sequencing the HVI and HVII regions? Forensic Sci. Int. 2000, 113, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Budowle, B.; Allard, M.W.; Wilson, M.R.; Chakraborty, R. Forensics and Mitochondrial DNA: Applications, Debates, and Foundations. Annu. Rev. Genom. Hum. Genet. 2003, 4, 119–141. [Google Scholar] [CrossRef]
- Ríos, L.; García-Rubio, A.; Martínez, B.; Alonso, A.; Puente, J. Identification process in mass graves from the Spanish Civil War II. Forensic Sci. Int. 2012, 219, e4–e9. [Google Scholar] [CrossRef] [PubMed]
- Piccinini, A.; Coco, S.; Parson, W.; Cattaneo, C.; Gaudio, D.; Barbazza, R.; Galassi, A. World War One Italian and Austrian soldier identification project: DNA results of the first case. Forensic Sci. Int. Genet. 2010, 4, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Ossowski, A.; Diepenbroek, M.; Kupiec, T.; Bykowska-Witowska, M.; Zielińska, G.; Dembińska, T.; Ciechanowicz, A. Genetic Identification of Communist Crimes’ Victims (1944–1956) Based on the Analysis of One of Many Mass Graves Discovered on the Powazki Military Cemetery in Warsaw, Poland. J. Forensic Sci. 2016, 61, 1450–1455. [Google Scholar] [CrossRef] [PubMed]
- Handt, O.; Richards, M.; Trommsdorff, M.; Kilger, C.; Simanainen, J.; Georgiev, O.; Bauer, K.; Stone, A.; Hedges, R.; Schaffner, W.; et al. Molecular genetic analyses of the Tyrolean Ice Man. Science 1994, 264, 1775–1778. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.J.; Li, Y.Z.; Yu, X.G.; Li, L.; Wu, D.Y.; Zhou, J.; Man, T.Y.; Yang, G.; Yan, J.W.; Cai, D.Q.; et al. Preliminary DNA identification for the tsunami victims in Thailand. Genom. Proteom. Bioinform. 2005, 3, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.; Montiel, R.; Sierra, B.; Bettencourt, C.; Fernandez, E.; Alvarez, L.; Lima, M.; Abade, A.; Aluja, M.P. Understanding Differences Between Phylogenetic and Pedigree-Derived mtDNA Mutation Rate: A Model Using Families from the Azores Islands (Portugal). Mol. Biol. Evol. 2005, 22, 1490–1505. [Google Scholar] [CrossRef]
- Sekiguchi, K.; Kasai, K.; Levin, B.C. Inter-and intragenerational transmission of a human mitochondrial DNA heteroplasmy among 13 maternally-related individuals and differences between and within tissues in two family members. Mitochondrion 2003, 2, 401–414. [Google Scholar] [CrossRef]
- Calloway, C.D.; Reynolds, R.L.; Herrin, G.L., Jr.; Anderson, W.W. The frequency of heteroplasmy in the HVII region of mtDNA differs across tissue types and increases with age. Am. J. Hum. Genet. 2000, 66, 1384–1397. [Google Scholar] [CrossRef] [PubMed]
- Bär, W.; Brinkmann, B.; Budowle, B.; Carracedo, A.; Gill, P.; Holland, M.; Lincoln, P.J.; Mayr, W.; Morling, N.; Olaisen, B.; et al. Guidelines for Mitochondrial DNA Typing. Vox Sang. 2000, 79, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Parson, W.; Gusmão, L.; Hares, D.R.; Irwin, J.A.; Mayr, W.R.; Morling, N.; Pokorak, E.; Prinz, M.; Salas, A.; Schneider, P.M.; et al. DNA Commission of the International Society for Forensic Genetics: Revised and extended guidelines for mitochondrial DNA typing. Forensic Sci. Int. Genet. 2014, 13, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.M.; Hopgood, R.; Gill, P. Identification of human remains by amplification and automated sequencing of mitochondrial DNA. Int. J. Legal Med. 1992, 105, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Handt, O.; Krings, M.; Ward, R.H.; Pääbo, S. The retrieval of ancient human DNA sequences. Am. J. Hum. Genet. 1996, 59, 368–376. [Google Scholar] [PubMed]
- Berger, C.; Parson, W. Mini-midi-mito: Adapting the amplification and sequencing strategy of mtDNA to the degradation state of crime scene samples. Forensic Sci. Int. Genet. 2009, 3, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.Y.; Lee, H.Y.; Park, S.J.; Yang, W.I.; Shin, K.-J. Modified Midi- and Mini-Multiplex PCR Systems for Mitochondrial DNA Control Region Sequence Analysis in Degraded Samples. J. Forensic Sci. 2013, 58, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Cooley, A.M. Mitochondrial DNA Analysis. In Forensic DNA Analysis: Methods and Protocols; Cupples Connon, C., Ed.; Springer: New York, NY, USA, 2023; pp. 331–349. [Google Scholar]
- Butler, J.M.; Coble, M.D.; Vallone, P.M. STRs vs. SNPs: Thoughts on the future of forensic DNA testing. Forensic Sci. Med. Pathol. 2007, 3, 200–205. [Google Scholar] [CrossRef]
- Andréasson, H.; Asp, A.; Alderborn, A.; Gyllensten, U.; Allen, M. Mitochondrial Sequence Analysis for Forensic Identification Using Pyrosequencing Technology. Biotechniques 2002, 32, 124–133. [Google Scholar] [CrossRef]
- Budowle, B. SNP Typing Strategies. Forensic Sci. Int. 2004, 146, S139–S142. [Google Scholar] [CrossRef]
- Novroski, N.M.M.; Cihlar, J.C. Evolution of single-nucleotide polymorphism use in forensic genetics. WIREs Forensic Sci. 2022, 4, e1459. [Google Scholar] [CrossRef]
- Sobrino, B.; Carracedo, A. SNP Typing in Forensic Genetics. In Forensic DNA Typing Protocols; Carracedo, A., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 107–126. [Google Scholar]
- Sanghavi, H. Recent Advancements in SNP Typing Methods Used in Forensic Science. In Advances in Genetic Polymorphisms; Nouha Bouayed, A., Balkiss, A., Eds.; IntechOpen: Rijeka, Yugoslavia, 2023; pp. 13–30. [Google Scholar]
- Budowle, B.; van Daal, A. Forensically relevant SNP classes. Biotechniques 2008, 44, 603–608. [Google Scholar] [CrossRef]
- Wendt, F.R.; Budowle, B. Pharmacogenetics and the postmortem molecular autopsy. WIREs Forensic Sci. 2020, 2, e1361. [Google Scholar] [CrossRef]
- Mehta, B.; Daniel, R.; Phillips, C.; McNevin, D. Forensically relevant SNaPshot® assays for human DNA SNP analysis: A review. Int. J. Leg. Med. 2017, 131, 21–37. [Google Scholar] [CrossRef]
- Tully, G.; Sullivan, K.M.; Nixon, P.; Stones, R.E.; Gill, P. Rapid Detection of Mitochondrial Sequence Polymorphisms Using Multiplex Solid-Phase Fluorescent Minisequencing. Genomics 1996, 34, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Glynn, C.L. Bridging Disciplines to Form a New One: The Emergence of Forensic Genetic Genealogy. Genes 2022, 13, 1381. [Google Scholar] [CrossRef]
- Ronaghi, M. Pyrosequencing sheds light on DNA sequencing. Genome Res. 2001, 11, 3–11. [Google Scholar] [CrossRef]
- Ahmadian, A.; Gharizadeh, B.; Gustafsson, A.C.; Sterky, F.; Nyrén, P.; Uhlén, M.; Lundeberg, J. Single-Nucleotide Polymorphism Analysis by Pyrosequencing. Anal. Biochem. 2000, 280, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, S.; Yamamoto, Y.; Doi, Y.; Takata, T.; Ishikawa, T.; Imabayashi, K.; Yoshitome, K.; Miyaishi, S.; Ishizu, H. A new 39-plex analysis method for SNPs including 15 blood group loci. Forensic Sci. Int. 2004, 144, 45–57. [Google Scholar] [CrossRef]
- Divne, A.-M.; Allen, M. A DNA microarray system for forensic SNP analysis. Forensic Sci. Int. 2005, 154, 111–121. [Google Scholar] [CrossRef]
- McNevin, D.; Bate, A.; Daniel, R.; Walsh, S.J. A preliminary mitochondrial DNA SNP genotyping assay for inferring genealogy. Aust. J. Forensic Sci. 2011, 43, 39–51. [Google Scholar] [CrossRef]
- Cornelis, S.; Gansemans, Y.; Deleye, L.; Deforce, D.; Van Nieuwerburgh, F. Forensic SNP Genotyping using Nanopore MinION Sequencing. Sci. Rep. 2017, 7, 41759. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xue, J.; Tan, M.; Chen, D.; Xiao, Y.; Liu, G.; Zheng, Y.; Wu, Q.; Liao, M.; Lv, M.; et al. An MPS-Based 50plex Microhaplotype Assay for Forensic DNA Analysis. Genes 2023, 14, 865. [Google Scholar] [CrossRef] [PubMed]
- Don, R.H.; Cox, P.T.; Wainwright, B.J.; Baker, K.; Mattick, J.S. ‘Touchdown’ PCR to circumvent spurious priming during gene amplification. Nucleic Acids Res. 1991, 19, 4008. [Google Scholar] [CrossRef]
- Coyle, P.V.; Ong, G.M.; O’Neill, H.J.; McCaughey, C.; De Ornellas, D.; Mitchell, F.; Mitchell, S.J.; Feeney, S.A.; Wyatt, D.E.; Forde, M.; et al. A touchdown nucleic acid amplification protocol as an alternative to culture backup for immunofluorescence in the routine diagnosis of acute viral respiratory tract infections. BMC Microbiol. 2004, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Sambrook, J. Touchdown Polymerase Chain Reaction (PCR). Cold Spring Harb. Protoc. 2018, 2018, pdb-prot095133. [Google Scholar] [CrossRef] [PubMed]
- Korbie, D.J.; Mattick, J.S. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nat. Protoc. 2008, 3, 1452–1456. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Won, J.; Choi, B.Y.; Lee, C.J. Optimization of primer sets and detection protocols for SARS-CoV-2 of coronavirus disease 2019 (COVID-19) using PCR and real-time PCR. Exp. Mol. Med. 2020, 52, 963–977. [Google Scholar] [CrossRef] [PubMed]
- Prezioso, V.R.; Jahns, A. Using Gradient PCR to Determine the Optimum Annealing Temperature; Eppendorf Scientific: Hamburg, Germany, 2000. [Google Scholar]
- ThermoFischer Scientific. PCR Thermal Cyclers Education. Available online: https://www.thermofisher.com/au/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-thermal-cyclers.html#:~:text=Peltier%20block%20elements%20are%20a,and%20better%20predict%20sample%20temperatures (accessed on 18 September 2023).
- Liu, H.; Fang, Y.; Su, X.; Wang, Y.; Ji, M.; Xing, H.; Gao, Y.; Zhang, Y.; He, N. Temperature control algorithm for polymerase chain reaction (PCR) instrumentation based upon improved hybrid fuzzy proportional integral derivative (PID) control. Instrum. Sci. Technol. 2023, 51, 109–131. [Google Scholar] [CrossRef]
- Nilsson, M.; Grånemo, J.; Buś, M.M.; Havsjö, M.; Allen, M. Comparison of DNA polymerases for improved forensic analysis of challenging samples. Forensic Sci. Int. Genet. 2016, 24, 55–59. [Google Scholar] [CrossRef]
- Yamagami, T.; Ishino, S.; Kawarabayasi, Y.; Ishino, Y. Mutant Taq DNA polymerases with improved elongation ability as a useful reagent for genetic engineering. Front. Microbiol. 2014, 5, 108670. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Kermekchiev, M.B.; Barnes, W.M. Direct DNA amplification from crude clinical samples using a PCR enhancer cocktail and novel mutants of Taq. J. Mol. Diagn. 2010, 12, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Dognaux, S.; Larmuseau, M.H.D.; Jansen, L.; Heylen, T.; Vanderheyden, N.; Bekaert, B.; Noel, F.; Decorte, R. Allele frequencies for the new European Standard Set (ESS) loci and D1S1677 in the Belgian population. Forensic Sci. Int. Genet. 2012, 6, e75–e77. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.M.; Hill, C.R.; Kline, M.C.; Bastisch, I.; Weirich, V.; McLaren, R.S.; Storts, D.R. SE33 variant alleles: Sequences and implications. Forensic Sci. Int. Genet. Suppl. Ser. 2011, 3, e502–e503. [Google Scholar] [CrossRef]
- Davis, C.; Ge, J.; King, J.; Malik, N.; Weirich, V.; Eisenberg, A.J.; Budowle, B. Variants observed for STR locus SE33: A concordance study. Forensic Sci. Int. Genet. 2012, 6, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Dixit, S.; Shrivastava, P.; Dash, H.R.; Kaitholia, K.; Sahajpal, V.; Sahoo, S.; Srivastava, V.; Surekha Rani, H.; Mishra, A.; Choudhary, S.K.; et al. Assessment of significance and forensic relevance of SE33 (ACTBP2) locus in five Indian populations. Gene Rep. 2021, 24, 101293. [Google Scholar] [CrossRef]
- Karunanathie, H.; Kee, P.S.; Ng, S.F.; Kennedy, M.A.; Chua, E.W. PCR enhancers: Types, mechanisms, and applications in long-range PCR. Biochimie 2022, 197, 130–143. [Google Scholar] [CrossRef]
- Klenow, H.; Henningsen, I. Effect of monovalent cations on the activity of the DNA polymerase of Escherichia coli B. Eur. J. Biochem. 1969, 9, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Balding, D.J.; Buckleton, J. Interpreting low template DNA profiles. Forensic Sci. Int. Genet. 2009, 4, 1–10. [Google Scholar] [CrossRef]
- Balding, D.J. Evaluation of mixed-source, low-template DNA profiles in forensic science. Proc. Natl. Acad. Sci. USA 2013, 110, 12241–12246. [Google Scholar] [CrossRef]
- Sidstedt, M.; Steffen, C.R.; Kiesler, K.M.; Vallone, P.M.; Rådström, P.; Hedman, J. The impact of common PCR inhibitors on forensic MPS analysis. Forensic Sci. Int. Genet. 2019, 40, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Akane, A.; Matsubara, K.; Nakamura, H.; Takahashi, S.; Kimura, K. Identification of the heme compound copurified with deoxyribonucleic acid (DNA) from bloodstains, a major inhibitor of polymerase chain reaction (PCR) amplification. J. Forensic Sci. 1994, 39, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Al-Soud, W.A.; Rådström, P. Purification and characterization of PCR-inhibitory components in blood cells. J. Clin. Microbiol. 2001, 39, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Sidstedt, M.; Hedman, J.; Romsos, E.L.; Waitara, L.; Wadsö, L.; Steffen, C.R.; Vallone, P.M.; Rådström, P. Inhibition mechanisms of hemoglobin, immunoglobulin G, and whole blood in digital and real-time PCR. Anal. Bioanal. Chem. 2018, 410, 2569–2583. [Google Scholar] [CrossRef] [PubMed]
- Bickley, J.; Short, J.K.; McDowell, D.G.; Parkes, H.C. Polymerase chain reaction (PCR) detection of Listeria monocytogenes in diluted milk and reversal of PCR inhibition caused by calcium ions. Lett. Appl. Microbiol. 1996, 22, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Rossen, L.; Nørskov, P.; Holmstrøm, K.; Rasmussen, O.F. Inhibition of PCR by components of food samples, microbial diagnostic assays and DNA-extraction solutions. Int. J. Food Microbiol. 1992, 17, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Schrader, C.; Schielke, A.; Ellerbroek, L.; Johne, R. PCR inhibitors—Occurrence, properties and removal. J. App. Microb. 2012, 113, 1014–1026. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, H.; Xu, Y.; Laššáková, S.; Korabečná, M.; Neužil, P. PCR past, present and future. Biotechniques 2020, 69, 317–325. [Google Scholar] [CrossRef]
- Berthier, J. Chapter 8—Biological Applications of EWOD. In Micro-Drops and Digital Microfluidics, 2nd ed.; Berthier, J., Ed.; William Andrew Publishing: Norwich, NY, USA, 2013; pp. 339–366. [Google Scholar]
- Brassard, D.; Malic, L.; Miville-Godin, C.; Normandin, F.; Veres, T. Advanced EWOD-based digital microfluidic system for multiplexed analysis of biomolecular interactions. In Proceedings of the 2011 IEEE 24th International Conference on Micro Electro Mechanical Systems, Cancun, Mexico, 23–27 January 2011; pp. 153–156. [Google Scholar]
- Schwarz, M.A.; Hauser, P.C. Recent developments in detection methods for microfabricated analytical devices. Lab A Chip 2001, 1, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Dittrich, P.S.; Manz, A. Single-molecule fluorescence detection in microfluidic channels-the Holy Grail in μtAS? Anal. Bioanal. Chem. 2005, 382, 1771–1782. [Google Scholar] [CrossRef]
- Luo, Y.; Bhattacharya, B.B.; Ho, T.Y.; Chakrabarty, K. Design and Optimization of a Cyberphysical Digital-Microfluidic Biochip for the Polymerase Chain Reaction. IEEE Trans. Comput. Aided Des. Integr. Circuits Syst. 2015, 34, 29–42. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, B.; Wu, W. Application of automatic feedback photographing by portable smartphone in PCR. Sens. Actuators B Chem. 2019, 298, 126782. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, L.; Guo, P.; Zhang, Y.; Lan, X.; Xu, W. Research progress of All-in-One PCR tube biosensors based on functional modification and intelligent fabrication. Biosens. Bioelectron. 2024, 246, 115824. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Brisk, P. Simulation of feedback-driven PCR assays on a 2D electrowetting array using a domain-specific high-level biological programming language. Microelectron. Eng. 2015, 148, 110–116. [Google Scholar] [CrossRef]
- Hua, Z.; Rouse, J.L.; Eckhardt, A.E.; Srinivasan, V.; Pamula, V.K.; Schell, W.A.; Benton, J.L.; Mitchell, T.G.; Pollack, M.G. Multiplexed real-time polymerase chain reaction on a digital microfluidic platform. Anal. Chem. 2010, 82, 2310–2316. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Yu, C.; Li, S.; Meng, J.; Li, T.; Cheng, J.; Li, J. A droplet-based multivolume microfluidic device for digital polymerase chain reaction. Sens. Actuators B Chem. 2022, 371, 132473. [Google Scholar] [CrossRef]
- McDonald, C.; Taylor, D.; Linacre, A. Towards a smart PCR process. Aust. J. Forensic Sci. 2023, 1–12. [Google Scholar] [CrossRef]
- McDonald, C.; Taylor, D.; Linacre, A. Smart PCR leading to improved DNA profiles. Aust. J. Forensic Sci. 2023. [Google Scholar] [CrossRef]

| Protocol | Year | Cycling Conditions | Total Cycles | Reaction Volume | ||
|---|---|---|---|---|---|---|
| Denaturation | Annealing | Extension | ||||
| Stoneking et al. [16] | 1991 | 94 °C 45 s | 56 °C 1 min | 74 °C 1 min | 30 | 100 L |
| Sullivan et al. [121] | 1992 | 94 °C 45 s | 50 °C 1 min | 72 °C 3 min | 32 | 25 L |
| Handt et al. [122] | 1996 | 92 °C 50 s | N/A 50 s | 72 °C 50 s | 40 | 40 L |
| Berger and Parson [123] | 2009 | 95 °C 15 s | 57 °C 10 s | 72 °C 20 s | 39 | 40 L |
| Kim et al. [124] | 2013 | 95 °C 20 s | 55 °C 60 s | 72 °C 30 s | 42 | 25 L |
| Cooley [125] | 2023 | 94 °C 20 s | 56 °C 20 s | 72 °C 30 s | 38 | 40 L |
| Protocol | Year | SNP Type | Cycling Conditions | Total Cycles | Reaction Volume | ||
|---|---|---|---|---|---|---|---|
| Denaturation | Annealing | Extension | |||||
| Tully et al. [135] | 1996 | Mitochondrial | 94 °C 30 s | 57 °C 30 s | 72 °C 90 s | 35 | 50 L |
| Ahmadian et al. [138] | 2000 | Nuclear | 94 °C 1 min | 50 °C 40 s | 72 °C 1 min | 35 | 50 L |
| Andréasson et al. [127] | 2001 | Mitochondrial | 95 °C 30 s | 54 °C or 60 °C 45 s | 72 °C 1 min | 40 | 100 L |
| Inagaki et al. [139] | 2004 | Nuclear | 96 °C 10 s | 50 °C 5 s | 60 °C 30 s | 25 | 5 L |
| Divne and Allen [140] | 2005 | Nuclear and Mitochondrial | 94 °C 30 s | 55 °C 30 s | 72 °C 30 s | 35 | 50 L |
| McNevin et al. [141] | 2011 | Mitochondrial | 94 °C 1 min | 56 °C 1 min | 72 °C 1 min | 30 | 25 L |
| STR Kit | Year | Loci Targeted | Total |
|---|---|---|---|
| PowerPlex1.1 [97] | 1997 | Amelogenin, CSF1PO, D5S818, D7S820, D13S317, D16S539, TH01, TPOX, vWA | 9 |
| PowerPlex 2.1 [98] | 1997 | D3S1358, D8S1179, D18S51, D21S11, FGA, Penta E, TH01, TPOX, vWA | 9 |
| SGM Plus [99] | 1999 | Amelogenin, D2S1338, D3S1358, D8S1179, D16S539, D18S51, D21S11, D19S433, FGA, TH01, vWA | 11 |
| PowerPlex 16 [100] | 2001 | Amelogenin, CSF1PO, D3S1358, D5S818, D7S820, D8S1179, D13S317, D16S539, D18S51, D21S11, FGA, Penta E, Penta D, TH01, TPOX, vWA | 16 |
| MiniFiler [102] | 2007 | Amelogenin, CSF1PO, D7S820, D13S317, D16S539, D18S51, D21S11, D2S1338, FGA | 9 |
| AmpFSTR Identifiler Plus [103] | 2010 | Amelogenin, CSF1PO, D2S1338, D3S1358, D5S818, D7S820, D8S1179, D13S317, D16S539, D18S51, D19S433, D21S11, FGA, TH01, TPOX, vWA | 16 |
| PowerPlex 21 [32] | 2012 | Amelogenin, CSF1PO, D1S1656, D2S1338, D3S1358, D5S818, D6S1043, D7S820, D8S1179, D12S391, D13S317, D16S539, D18S51, D19S433, D21S11, FGA, Penta D, Penta E, TH01, TPOX, vWA | 21 |
| GlobalFiler and GlobalFiler IQC [35] | 2013 | Amelogenin, CSF1PO, D1S1656, D2S441, D2S1338, D3S1358, D5S818, D7S820, D8S1179, D10S1248, D12S391, D13S317, D16S539, D18S51, D19S433, D21S11, D22S1045, DYS391, FGA, TH01, TPOX, SE33, vWA, Yindel | 24 |
| VeriFiler Plus [106] | 2018 | Amelogenin, CSF1PO, D1S1656, D2S441, D2S1338, D3S1358, D5S818, D6S1043, D7S820, D8S1179, D10S1248, D12S391, D13S317, D16S539, D18S51, D19S433, D21S11, D22S1045, FGA, Penta E, Penta D, TH01, TPOX, vWA, Yindel | 25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McDonald, C.; Taylor, D.; Linacre, A. PCR in Forensic Science: A Critical Review. Genes 2024, 15, 438. https://doi.org/10.3390/genes15040438
McDonald C, Taylor D, Linacre A. PCR in Forensic Science: A Critical Review. Genes. 2024; 15(4):438. https://doi.org/10.3390/genes15040438
Chicago/Turabian StyleMcDonald, Caitlin, Duncan Taylor, and Adrian Linacre. 2024. "PCR in Forensic Science: A Critical Review" Genes 15, no. 4: 438. https://doi.org/10.3390/genes15040438
APA StyleMcDonald, C., Taylor, D., & Linacre, A. (2024). PCR in Forensic Science: A Critical Review. Genes, 15(4), 438. https://doi.org/10.3390/genes15040438