
Performance of a 74-Microhaplotype Assay in Kinship Analyses

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. DNA Extraction, DNA Quantification, and PCR Amplification
2.3. Library Building and DNA Sequencing
2.4. Haplotype Calling
2.5. Population Genetic Analyses
2.6. Simulations and LR Calculations
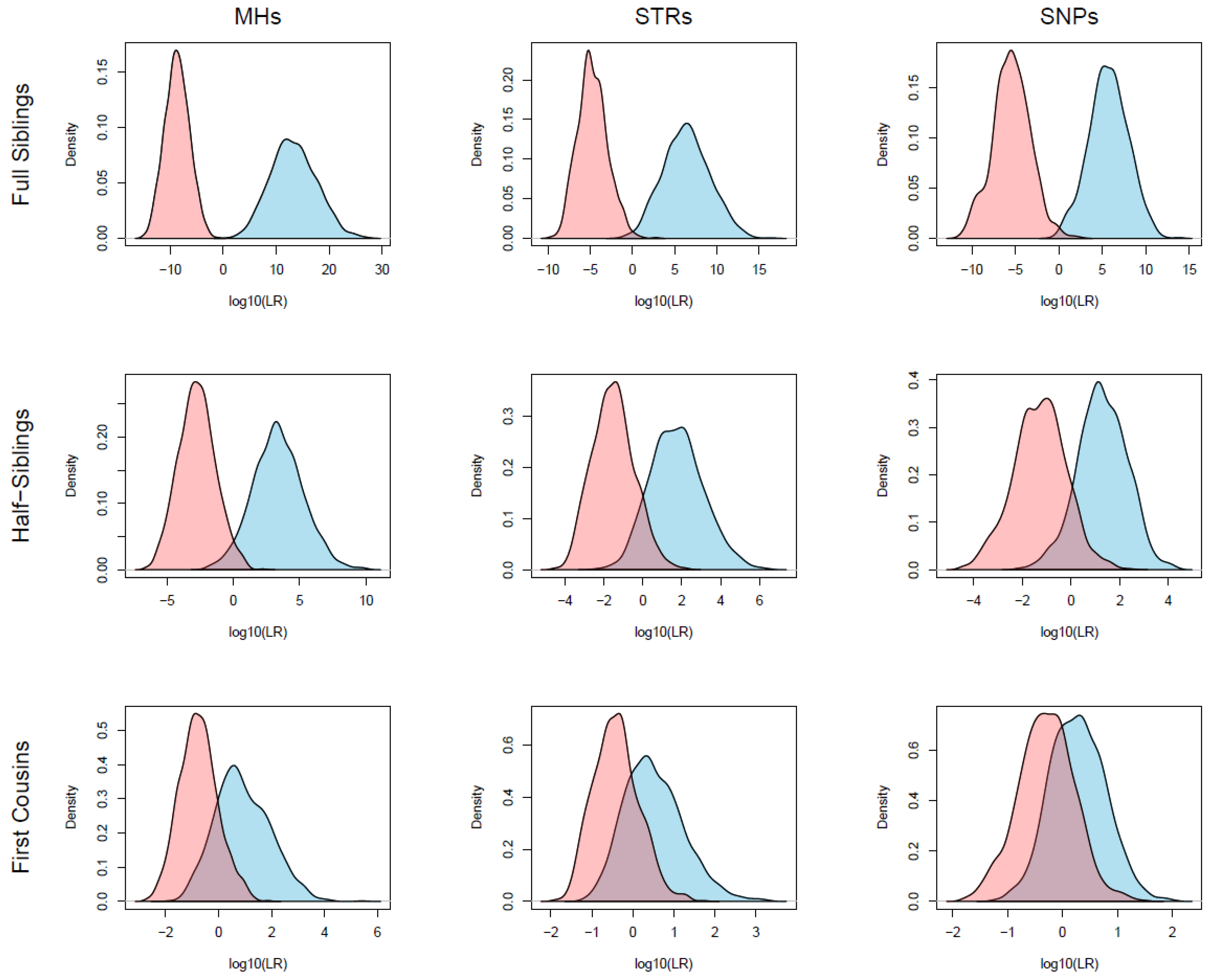
3. Results
3.1. Sequencing Results and Haplotype Calling
3.2. Population Genetics Parameters
3.3. Kinship Simulations and LR Calculations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gill, P.; Haned, H.; Bleka, O.; Hansson, O.; Dørum, G.; Egeland, T. Genotyping and Interpretation of STR-DNA: Low-Template, Mixtures and Database Matches—Twenty Years of Research and Development. Forensic Sci. Int. Genet. 2015, 18, 100–117. [Google Scholar] [CrossRef]
- Børsting, C.; Morling, N. Next Generation Sequencing and Its Applications in Forensic Genetics. Forensic Sci. Int. Genet. 2015, 18, 78–89. [Google Scholar] [CrossRef]
- Kidd, K.K.; Pakstis, A.J.; Speed, W.C.; Lagacé, R.; Chang, J.; Wootton, S.; Haigh, E.; Kidd, J.R. Current Sequencing Technology Makes Microhaplotypes a Powerful New Type of Genetic Marker for Forensics. Forensic Sci. Int. Genet. 2014, 12, 215–224. [Google Scholar] [CrossRef]
- Oldoni, F.; Kidd, K.K.; Podini, D. Microhaplotypes in Forensic Genetics. Forensic Sci. Int. Genet. 2019, 38, 54–69. [Google Scholar] [CrossRef]
- Ellegren, H. Microsatellite Mutations in the Germline: Implications for Evolutionary Inference. Trends Genet. TIG 2000, 16, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Fung, W.K.; Wong, D.; Hu, Y. Full Siblings Impersonating Parent/Child Prove Most Difficult to Discredit with DNA Profiling Alone. Transfusion 2004, 44, 1513–1515. [Google Scholar] [CrossRef] [PubMed]
- von Wurmb-Schwark, N.; Mályusz, V.; Simeoni, E.; Lignitz, E.; Poetsch, M. Possible Pitfalls in Motherless Paternity Analysis with Related Putative Fathers. Forensic Sci. Int. 2006, 159, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.; Fondevila, M.; García-Magariños, M.; Rodriguez, A.; Salas, A.; Carracedo, A.; Lareu, M.V. Resolving Relationship Tests That Show Ambiguous STR Results Using Autosomal SNPs as Supplementary Markers. Forensic Sci. Int. Genet. 2008, 2, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Hussing, C.; Huber, C.; Bytyci, R.; Mogensen, H.S.; Morling, N.; Børsting, C. Sequencing of 231 Forensic Genetic Markers Using the MiSeq FGxTM Forensic Genomics System—An Evaluation of the Assay and Software. Forensic Sci. Res. 2018, 3, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Kidd, K.K.; Speed, W.C.; Pakstis, A.J.; Podini, D.S.; Lagacé, R.; Chang, J.; Wootton, S.; Haigh, E.; Soundararajan, U. Evaluating 130 Microhaplotypes across a Global Set of 83 Populations. Forensic Sci. Int. Genet. 2017, 29, 29–37. [Google Scholar] [CrossRef] [PubMed]
- De La Puente, M.; Phillips, C.; Xavier, C.; Amigo, J.; Carracedo, A.; Parson, W.; Lareu, M.V. Building a Custom Large-Scale Panel of Novel Microhaplotypes for Forensic Identification Using MiSeq and Ion S5 Massively Parallel Sequencing Systems. Forensic Sci. Int. Genet. 2020, 45, 102213. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Liu, Y.; Li, J.; Yang, Z.; Wen, D.; Liang, W.; Yan, Y.; Yu, H.; Cai, J.; Zha, L. Development and Application of a Nonbinary SNP-Based Microhaplotype Panel for Paternity Testing Involving Close Relatives. Forensic Sci. Int. Genet. 2020, 46, 102255. [Google Scholar] [CrossRef] [PubMed]
- Gandotra, N.; Speed, W.C.; Qin, W.; Tang, Y.; Pakstis, A.J.; Kidd, K.K.; Scharfe, C. Validation of Novel Forensic DNA Markers Using Multiplex Microhaplotype Sequencing. Forensic Sci. Int. Genet. 2020, 47, 102275. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Li, H.; Li, R.; Peng, D.; Wang, N.; Shen, X.; Sun, H. Identification and Sequencing of 59 Highly Polymorphic Microhaplotypes for Analysis of DNA Mixtures. Int. J. Leg. Med. 2021, 135, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.-S.; Feng, Y.-S.; Kang, K.-L.; Zhang, C.; Ji, A.-Q.; Ye, J.; Wang, L. Screening of Highly Discriminative Microhaplotype Markers for Individual Identification and Mixture Deconvolution in East Asian Populations. Forensic Sci. Int. Genet. 2022, 59, 102720. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Ma, G.; Lu, C.; Wang, Q.; Fu, L.; Cong, B.; Li, S. Development and Evaluation of a Novel Panel Containing 188 Microhaplotypes for 2nd-Degree Kinship Testing in the Hebei Han Population. Forensic Sci. Int. Genet. 2023, 65, 102855. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Wang, H.; Cao, Y.; Huang, Y.; Wei, Y.; Hu, Y.; Dai, X.; Shan, T.; Wang, Y.; Zhang, J. Evaluation of Large-Scale Highly Polymorphic Microhaplotypes in Complex DNA Mixtures Analysis Using RMNE Method. Forensic Sci. Int. Genet. 2023, 65, 102874. [Google Scholar] [CrossRef] [PubMed]
- The 1000 Genomes Project Consortium. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Turchi, C.; Melchionda, F.; Pesaresi, M.; Tagliabracci, A. Evaluation of a Microhaplotypes Panel for Forensic Genetics Using Massive Parallel Sequencing Technology. Forensic Sci. Int. Genet. 2019, 41, 120–127. [Google Scholar] [CrossRef]
- Oldoni, F.; Bader, D.; Fantinato, C.; Wootton, S.C.; Lagacé, R.; Kidd, K.K.; Podini, D. A Sequence-Based 74plex Microhaplotype Assay for Analysis of Forensic DNA Mixtures. Forensic Sci. Int. Genet. 2020, 49, 102367. [Google Scholar] [CrossRef]
- Wu, R.; Chen, H.; Li, R.; Zang, Y.; Shen, X.; Hao, B.; Wang, Q.; Sun, H. Pairwise Kinship Testing with Microhaplotypes: Can Advancements Be Made in Kinship Inference with These Markers? Forensic Sci. Int. 2021, 325, 110875. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.-B.; Rao, M.; Chen, Q.-F.; Ji, A.-Q.; Zhang, C.; Kang, K.-L.; Wu, H.; Ye, J.; Nie, S.-J.; Wang, L. A 124-Plex Microhaplotype Panel Based on Next-Generation Sequencing Developed for Forensic Applications. Sci. Rep. 2020, 10, 1945. [Google Scholar] [CrossRef] [PubMed]
- Staadig, A.; Tillmar, A. Evaluation of Microhaplotypes in Forensic Kinship Analysis from a Swedish Population Perspective. Int. J. Leg. Med. 2021, 135, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Crow, J.F.; Kimura, M. An Introduction to Population Genetics Theory; Harper & Row: New York, NY, USA, 1970. [Google Scholar]
- Rosenberg, N.A.; Li, L.M.; Ward, R.; Pritchard, J.K. Informativeness of Genetic Markers for Inference of Ancestry. Am. J. Hum. Genet. 2003, 73, 1402–1422. [Google Scholar] [CrossRef] [PubMed]
- Kidd, K.K.; Pakstis, A.J.; Speed, W.C.; Lagace, R.; Wootton, S.; Chang, J. Selecting Microhaplotypes Optimized for Different Purposes. Electrophoresis 2018, 39, 2815–2823. [Google Scholar] [CrossRef]
- Jønck, C.G.; Børsting, C. Introduction of the Python Script MHinNGS for Analysis of Microhaplotypes. Forensic Sci. Int. Genet. Suppl. Ser. 2022, 8, 79–81. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin Suite Ver 3.5: A New Series of Programs to Perform Population Genetics Analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Holm, S. A Simple Sequentially Rejective Multiple Test Procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]
- Abecasis, G.R.; Cherny, S.S.; Cookson, W.O.; Cardon, L.R. Merlin–Rapid Analysis of Dense Genetic Maps Using Sparse Gene Flow Trees. Nat. Genet. 2002, 30, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Tomas, C.; Mogensen, H.S.; Friis, S.L.; Hallenberg, C.; Stene, M.C.; Morling, N. Concordance Study and Population Frequencies for 16 Autosomal STRs Analyzed with PowerPlex® ESI 17 and AmpFℓSTR® NGM SElectTM in Somalis, Danes and Greenlanders. Forensic Sci. Int. Genet. 2014, 11, e18–e21. [Google Scholar] [CrossRef]
- Pereira, V.; Tomas, C.; Sanchez, J.J.; Syndercombe-Court, D.; Amorim, A.; Gusmão, L.; Prata, M.J.; Morling, N. The Peopling of Greenland: Further Insights from the Analysis of Genetic Diversity Using Autosomal and X-Chromosomal Markers. Eur. J. Hum. Genet. 2015, 23, 245–251. [Google Scholar] [CrossRef]
- Buchard, A.; Kampmann, M.; Poulsen, L.; Børsting, C.; Morling, N. ISO 17025 Validation of a Next-generation Sequencing Assay for Relationship Testing. Electrophoresis 2016, 37, 2822–2831. [Google Scholar] [CrossRef]
- Sanchez, J.J.; Phillips, C.; Børsting, C.; Balogh, K.; Bogus, M.; Fondevila, M.; Harrison, C.D.; Musgrave-Brown, E.; Salas, A.; Syndercombe-Court, D.; et al. A Multiplex Assay with 52 Single Nucleotide Polymorphisms for Human Identification. Electrophoresis 2006, 27, 1713–1724. [Google Scholar] [CrossRef] [PubMed]
- Nato, A.Q.; Buyske, S.; Matise, T.C. The Rutgers Map: A Third-Generation Combined Linkage-Physical Map of the Human Genome. Human Genetics Institute of New Jersey Second Research Day; Life Sciences Building, Rutgers University: Piscataway, NJ, USA, 2018; Available online: http://compgen.rutgers.edu/rutgers_maps.shtml (accessed on 8 January 2024).
- Jønck, C.G.; Qian, X.; Simayijiang, H.; Børsting, C. STRinNGS v2.0: Improved Tool for Analysis and Reporting of STR Sequencing Data. Forensic Sci. Int. Genet. 2020, 48, 102331. [Google Scholar] [CrossRef]
- Tomas, C.; Pereira, V.; Morling, N. Analysis of 12 X-STRs in Greenlanders, Danes and Somalis Using Argus X-12. Int. J. Leg. Med. 2012, 126, 121–128. [Google Scholar] [CrossRef]
- Moltke, I.; Fumagalli, M.; Korneliussen, T.S.; Crawford, J.E.; Bjerregaard, P.; Jørgensen, M.E.; Grarup, N.; Gulløv, H.C.; Linneberg, A.; Pedersen, O.; et al. Uncovering the Genetic History of the Present-Day Greenlandic Population. Am. J. Hum. Genet. 2015, 96, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, J.K.; Pereira, V.; Børsting, C.; Morling, N. Peopling of the North Circumpolar Region—Insights from Y Chromosome STR and SNP Typing of Greenlanders. PLoS ONE 2015, 10, e0116573. [Google Scholar] [CrossRef] [PubMed]
- Lopopolo, M.; Børsting, C.; Pereira, V.; Morling, N. A Study of the Peopling of Greenland Using next Generation Sequencing of Complete Mitochondrial Genomes. Am. J. Biol. Anthropol. 2016, 161, 698–704. [Google Scholar] [CrossRef]
- Tillmar, A.; Sturk-Andreaggi, K.; Daniels-Higginbotham, J.; Thomas, J.T.; Marshall, C. The FORCE Panel: An All-in-One SNP Marker Set for Confirming Investigative Genetic Genealogy Leads and for General Forensic Applications. Genes 2021, 12, 1968. [Google Scholar] [CrossRef]
- Staadig, A.; Tillmar, A. An Overall Limited Effect on the Weight-of-Evidence When Taking STR DNA Sequence Polymorphism into Account in Kinship Analysis. Forensic Sci. Int. Genet. 2019, 39, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.M.; Mogensen, H.S.; Eriksen, P.S.; Olofsson, J.K.; Asplund, M.; Morling, N. Estimating Y-STR Allelic Drop-out Rates and Adjusting for Interlocus Balances. Forensic Sci. Int. Genet. 2013, 7, 327–336. [Google Scholar] [CrossRef]
- Pereira, V.; Longobardi, A.; Børsting, C. Sequencing of Mitochondrial Genomes Using the Precision ID mtDNA Whole Genome Panel. Electrophoresis 2018, 39, 2766–2775. [Google Scholar] [CrossRef]
- Kling, D.; Egeland, T.; Tillmar, A.O. FamLink—A User Friendly Software for Linkage Calculations in Family Genetics. Forensic Sci. Int. Genet. 2012, 6, 616–620. [Google Scholar] [CrossRef] [PubMed]
- Standage, D.S.; Mitchell, R.N. MicroHapDB: A Portable and Extensible Database of All Published Microhaplotype Marker and Frequency Data. Front. Genet. 2020, 11, 781. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Marker Sets | Denmark | Greenland | ||||
|---|---|---|---|---|---|---|
| Full Siblings | Half-Siblings | First Cousins | Full Siblings | Half-Siblings | First Cousins | |
| MHs * | 1.87 × 1013 | 2412 | 8 | 4.62 × 108 | 147 | 4 |
| STRs ** | 2.55 × 106 | 57 | 3 | 2.04 × 105 | 30 | 3 |
| SNPs *** | 5.54 × 105 | 19 | 2 | 168 | 3 | 1 |
| Relationship | LR Threshold | MH | STR | SNP | |||
|---|---|---|---|---|---|---|---|
| False Positives † | False Negatives ‡ | False Positives † | False Negatives ‡ | False Positives † | False Negatives ‡ | ||
| Siblings | 1 | 0.00% | 0.00% | 0.70% | 0.80% | 1.00% | 0.20% |
| 10 | 0.00% | 0.00% | 0.20% | 1.70% | 0.40% | 2.20% | |
| 100 | 0.00% | 0.10% | 0.10% | 5.30% | 0.10% | 5.60% | |
| 1000 | 0.00% | 0.30% | 0.00% | 11.80% | 0.00% | 11.00% | |
| 10,000 | 0.00% | 1.10% | 0.00% | 18.80% | 0.00% | 21.10% | |
| Half-siblings | 1 | 3.10% | 3.60% | 8.80% | 9.40% | 11.40% | 10.00% |
| 10 | 0.30% | 10.00% | 1.30% | 30.90% | 2.00% | 38.30% | |
| 100 | 0.10% | 24.00% | 0.20% | 57.00% | 0.20% | 75.20% | |
| 1000 | 0.00% | 41.60% | 0.00% | 81.40% | 0.00% | 96.30% | |
| 10,000 | 0.00% | 63.90% | 0.00% | 94.10% | 0.00% | 99.60% | |
| Cousins | 1 | 14.40% | 18.30% | 22.70% | 26.00% | 27.70% | 31.00% |
| 10 | 1.00% | 56.60% | 1.40% | 76.10% | 1.10% | 92.20% | |
| 100 | 0.00% | 84.50% | 0.00% | 96.60% | 0.00% | 99.90% | |
| 1000 | 0.00% | 96.90% | 0.00% | 99.70% | 0.00% | 100.00% | |
| 10,000 | 0.00% | 99.70% | 0.00% | 100.00% | 0.00% | 100.00% | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomas, C.; Rodrigues, P.; Jønck, C.G.; Barekzay, Z.; Simayijiang, H.; Pereira, V.; Børsting, C. Performance of a 74-Microhaplotype Assay in Kinship Analyses. Genes 2024, 15, 224. https://doi.org/10.3390/genes15020224
Tomas C, Rodrigues P, Jønck CG, Barekzay Z, Simayijiang H, Pereira V, Børsting C. Performance of a 74-Microhaplotype Assay in Kinship Analyses. Genes. 2024; 15(2):224. https://doi.org/10.3390/genes15020224
Chicago/Turabian StyleTomas, Carmen, Pedro Rodrigues, Carina G. Jønck, Zohal Barekzay, Halimureti Simayijiang, Vania Pereira, and Claus Børsting. 2024. "Performance of a 74-Microhaplotype Assay in Kinship Analyses" Genes 15, no. 2: 224. https://doi.org/10.3390/genes15020224
APA StyleTomas, C., Rodrigues, P., Jønck, C. G., Barekzay, Z., Simayijiang, H., Pereira, V., & Børsting, C. (2024). Performance of a 74-Microhaplotype Assay in Kinship Analyses. Genes, 15(2), 224. https://doi.org/10.3390/genes15020224