
Exploring the Impact of Primer–Template Mismatches on PCR Performance of DNA Polymerases Varying in Proofreading Activity

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Establishment of a Chlamydia pneumonia FRET-PCR System
2.2. Nucleotide Mismatches in the Primers
2.3. Calculation of the Relative PCR Amplification Efficiency
2.4. Establishment of Babesia PCR to Differentiate Babesia and Theileria
3. Results
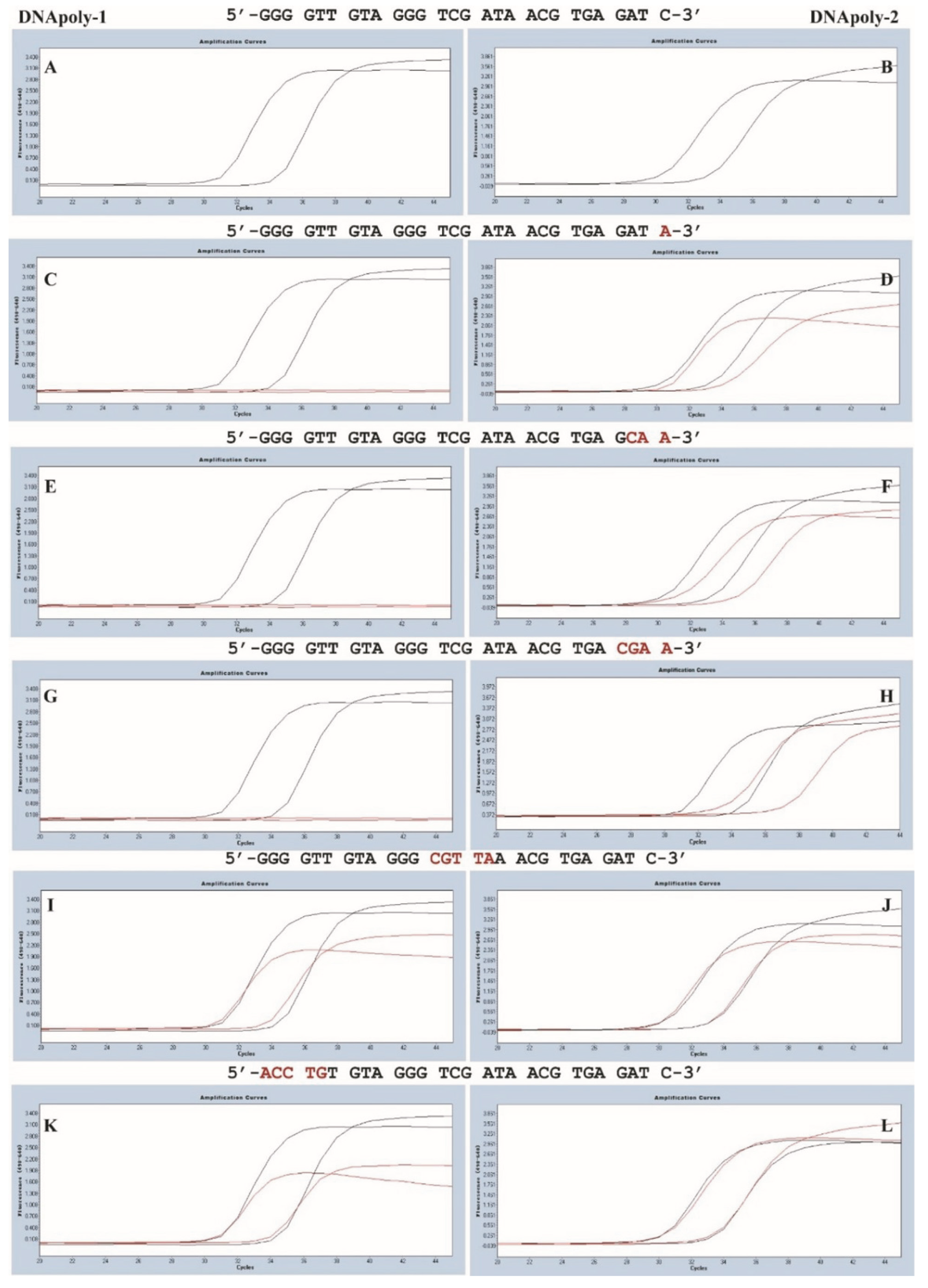
3.1. Establishment of a Highly Sensitive Quantitative PCR
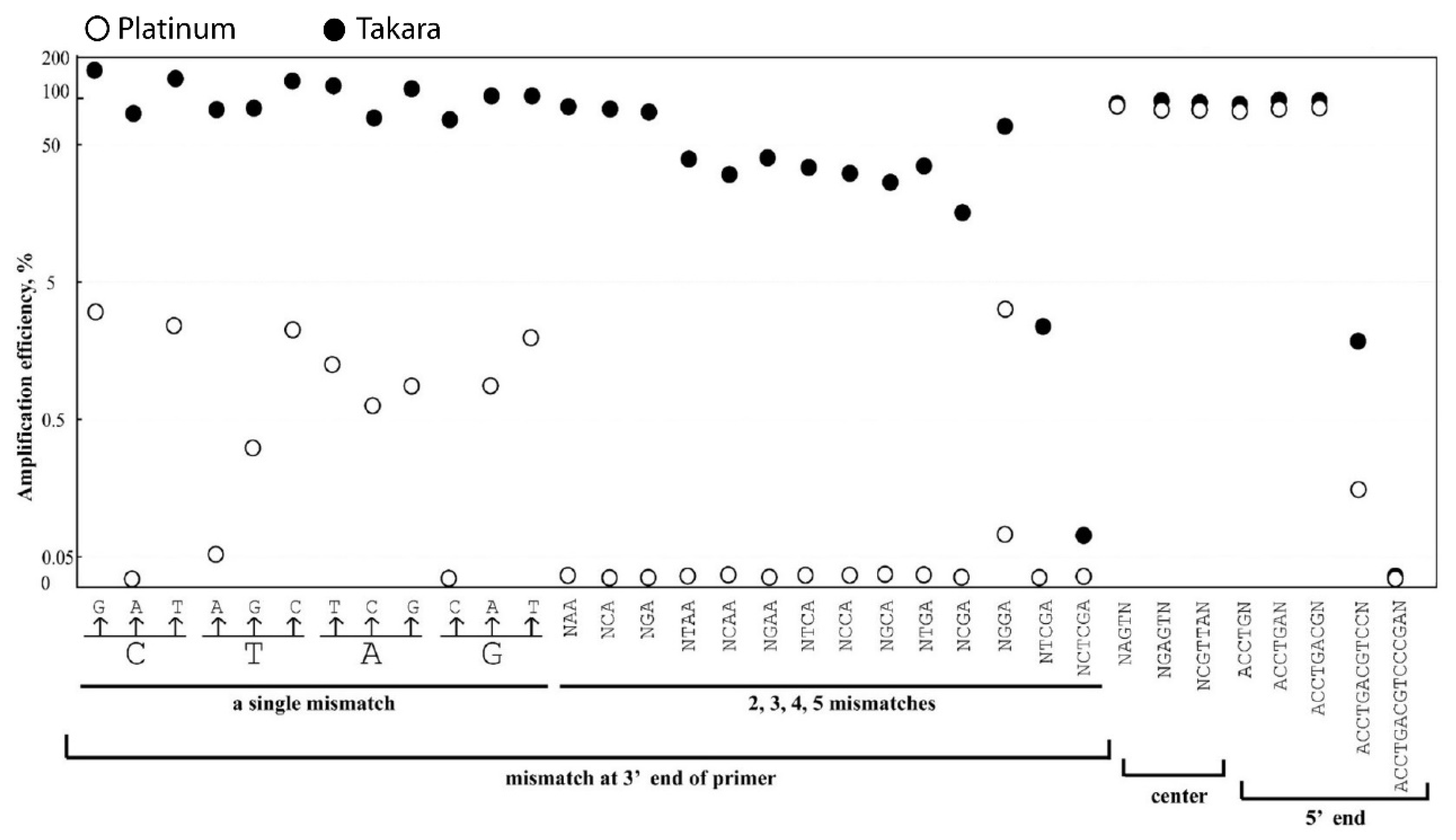
3.2. A Single-Nucleotide Mismatch at 3′ Primer Reduced the Amplification Efficiency When Platinum, but Not Takara, Was Used
3.3. Nucleotide Mismatch in the Center and 5’ End of the Primer Influences the Amplification Moderately
3.4. Amplification Efficiencies Affected by Nucleotide Mismatches in Upstream Primer, Center, and Downstream of the Primer
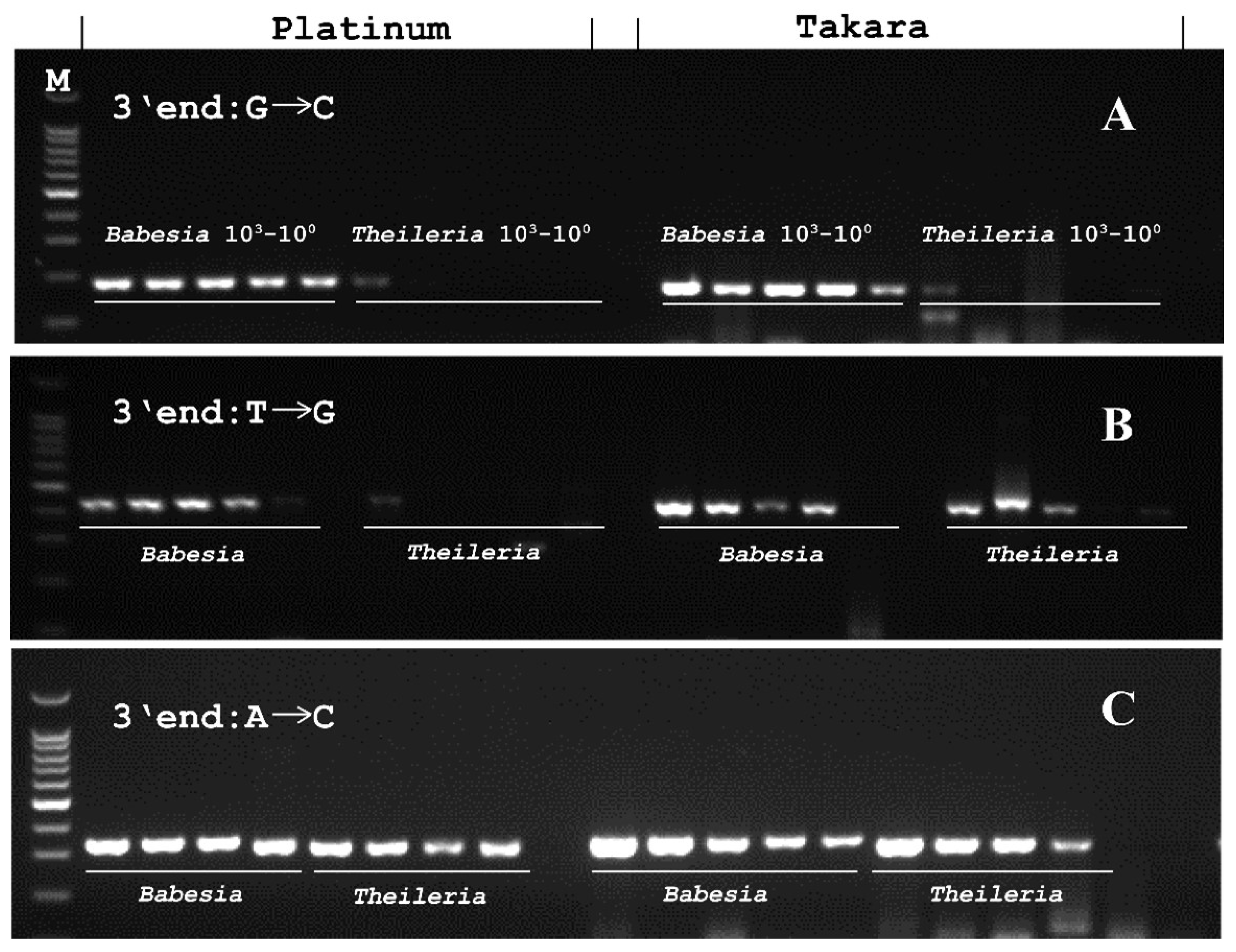
3.5. Designed Babesia PCR Specifically Amplifies Babesia, but Not Theileria
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saiki, R.K.; Scharf, S.; Faloona, F. Enzymatic amplification of Beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science 1985, 230, 1350–1354. [Google Scholar] [CrossRef]
- Saiki, R.K.; Gelfand, D.H.; Stoffel, S. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science 1988, 239, 487–491. [Google Scholar] [CrossRef]
- Mullis, K.B. The unusual origin of the polymerase chain reaction. Sci. Am. 1990, 262, 56–61+64–65. [Google Scholar] [CrossRef]
- Kaltenboeck, B.; Wang, C. Advances in real-time PCR: Application to clinical laboratory diagnostics. Adv. Clin. Chem. 2005, 40, 219–259. [Google Scholar] [PubMed]
- Farrell, R.E., Jr. DNA amplification. Immunol. Investig. 1997, 26, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Sachse, K. Specificity and performance of PCR detection assays for microbial pathogens. Mol. Biotechnol. 2004, 26, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Cline, J.; Braman, J.C.; Hogrefe, H.H. PCR fidelity of pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res. 1996, 24, 3546–3551. [Google Scholar] [CrossRef] [PubMed]
- Steitz, T.A. DNA polymerases: Structural diversity and common mechanisms. J. Biol. Chem. 1999, 274, 17395–17398. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.M.; Arnheim, N.; Goodman, M.F. Extension of base mispairs by Taq DNA polymerase: Implications for single nucleotide discrimination in PCR. Nucleic Acids Res. 1992, 20, 4567–4573. [Google Scholar] [CrossRef] [PubMed]
- Ayyadevara, S.; Thaden, J.J.; Shmookler Reis, R.J. Discrimination of primer 3′-nucleotide mismatch by taq DNA polymerase during polymerase chain reaction. Anal. Biochem. 2000, 284, 11–18. [Google Scholar] [CrossRef]
- Whiley, D.M.; Sloots, T.P. Sequence variation in primer targets affects the accuracy of viral quantitative PCR. J. Clin. Virol. 2005, 34, 104–107. [Google Scholar] [CrossRef]
- Miura, F.; Uematsu, C.; Sakaki, Y.; Ito, T. A novel strategy to design highly specific PCR primers based on the stability and uniqueness of 3′-end subsequences. Bioinformatics 2005, 21, 4363–4370. [Google Scholar] [CrossRef]
- Stadhouders, R.; Pas, S.D.; Anber, J.; Voermans, J.; Mes, T.H.; Schutten, M. The effect of primer-template mismatches on the detection and quantification of nucleic acids using the 5′ nuclease assay. J. Mol. Diagn. 2010, 12, 109–117. [Google Scholar] [CrossRef]
- Wu, J.H.; Hong, P.Y.; Liu, W.T. Quantitative effects of position and type of single mismatch on single base primer extension. J. Microbiol. Methods 2009, 77, 267–275. [Google Scholar] [CrossRef]
- Boyle, B.; Dallaire, N.; MacKay, J. Evaluation of the impact of single nucleotide polymorphisms and primer mismatches on quantitative PCR. BMC Biotechnol. 2009, 9, 75. [Google Scholar] [CrossRef]
- Lefever, S.; Pattyn, F.; Hellemans, J.; Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin. Chem. 2013, 59, 1470–1480. [Google Scholar] [CrossRef]
- Goodwin, J. Primer-Template Mismatch Does Not Contribute to Irreproducibility of PCR Replicates. J. Exp. Microbiol. Immunol. 2013, 17, 129–132. [Google Scholar]
- DeGraves, F.J.; Gao, D.; Kaltenboeck, B. High-sensitivity quantitative PCR platform. Biotechniques 2003, 34, 106–110. [Google Scholar] [CrossRef]
- Guo, W.; Li, J.; Kaltenboeck, B.; Gong, J.; Fan, W.; Wang, C. Chlamydia gallinacea, not C. psittaci, is the endemic chlamydial species in chicken (Gallus gallus). Sci. Rep. 2016, 6, 19638. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Olmeda, A.S.; Armstrong, P.M.; Rosenthal, B.M.; Valladares, B.; del Castillo, A.; de Armas, F.; Miguelez, M.; González, A.; Rodríguez Rodríguez, J.A.; Spielman, A.; et al. A subtropical case of human babesiosis. Acta Trop. 1997, 67, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Casati, S.; Sager, H.; Gern, L.; Piffaretti, J.C. Presence of potentially pathogenic Babesia sp. forhuman in Ixodes ricinus in Switzerland. Ann. Agric. Environ. Med. 2006, 13, 65–70. [Google Scholar]
- Drum, M.; Kranaster, R.; Ewald, C.; Blasczyk, R.; Marx, A. Variants of a Thermus aquaticus DNA polymerase with increased selectivity for applications in allele- and methylation-specific amplification. PLoS ONE 2014, 9, e96640. [Google Scholar] [CrossRef]
- Lim, Y.; Park, I.H.; Lee, H.H.; Baek, K.; Lee, B.C.; Cho, G. Modified Taq DNA Polymerase for Allele-Specific Ultra-Sensitive Detection of Genetic Variants. J. Mol. Diagn. 2022, 24, 1128–1142. [Google Scholar] [CrossRef]
- Pérez, L.M.; Fittipaldi, M.; Adrados, B.; Morató, J.; Codony, F. Error estimation in environmental DNA targets quantification due to PCR efficiencies differences between real samples and standards. Folia Microbiol. (Praha) 2013, 58, 657–662. [Google Scholar] [CrossRef]
- Ramakers, C.; Ruijter, J.M.; Deprez, R.H.; Moorman, A.F. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 2003, 339, 62–66. [Google Scholar] [CrossRef]
- Untergasser, A.; Ruijter, J.M.; Benes, V.; van den Hoff, M.J.B. Web-based LinRegPCR: Application for the visualization and analysis of (RT)-qPCR amplification and melting data. BMC Bioinform. 2021, 22, 398. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Nucleotide Mismatch | Amplification Efficiency | |
|---|---|---|
| GGGGTTGTAGGGTCGATAACGTGAGATC | Platinum, 100% | Takara, 100% |
| GGGGTTGTAGGGTCGATAACGTGAGATG * | 4% | 190% |
| GGGGTTGTAGGGTCGATAACGTGAGATA | 0% | 90% |
| GGGGTTGTAGGGTCGATAACGTGAGATT | 3% | 165% |
| GGGGTTGTAGGGTCGATAACGTGAGAA | 0% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGAG | 0% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGAC | 3% | 160% |
| GGGGTTGTAGGGTCGATAACGTGAGT | 1% | 150% |
| GGGGTTGTAGGGTCGATAACGTGAGC | 2% | 95% |
| GGGGTTGTAGGGTCGATAACGTGAGG | 1% | 130% |
| GGGGTTGTAGGGTCGATAACGTGAC | 0% | 80% |
| GGGGTTGTAGGGTCGATAACGTGAA | 1% | 115% |
| GGGGTTGTAGGGTCGATAACGTGAT | 2% | 120% |
| GGGGTTGTAGGGTCGATAACGTGAGATM ** | 59% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGATS | 56% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGATY | 63% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGATH | 48% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGATV | 39% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGATB | 43% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGATN | 34% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGAGC | 0% | 90% |
| GGGGTTGTAGGGTCGATAACGTGAGAAC | 1% | 93% |
| GGGGTTGTAGGGTCGATAACGTGAGACC | 13% | 95% |
| GGGGTTGTAGGGTCGATAACGTGAGCTC | 12% | 92% |
| GGGGTTGTAGGGTCGATAACGTGAGTTC | 12% | 92% |
| GGGGTTGTAGGGTCGATAACGTGAGGTC | 5% | 85% |
| GGGGTTGTAGGGTCGATAACGTGATATC | 37% | 98% |
| GGGGTTGTAGGGTCGATAACGTGCGATC | 82% | 100% |
| Nucleotide Mismatch | Amplification Efficiency | |
|---|---|---|
| GGGGTTGTAGGGTCGATAACGTGAGATC | Platinum, 100% | Takara, 100% |
| GGGGTTGTAGGGTCGATAACGTGAGAAG | 0% | 160% |
| GGGGTTGTAGGGTCGATAACGTGAGACG | 0% | 87% |
| GGGGTTGTAGGGTCGATAACGTGAGAGG | 0% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGAAA | 0% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGACA | 0% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGAGA | 0% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGAAT | 0% | 170% |
| GGGGTTGTAGGGTCGATAACGTGAGACT | 1% | 138% |
| GGGGTTGTAGGGTCGATAACGTGAGAGT | 0% | 150% |
| GGGGTTGTAGGGTCGATAACGTGAGTAG | 0% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGCAG | 0% | 73% |
| GGGGTTGTAGGGTCGATAACGTGAGGAG | 0% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGTCG | 0% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGCCG | 0% | 72% |
| GGGGTTGTAGGGTCGATAACGTGAGGCG | 0% | 99% |
| GGGGTTGTAGGGTCGATAACGTGAGTGG | 0% | 85% |
| GGGGTTGTAGGGTCGATAACGTGAGCGG | 0% | 43% |
| GGGGTTGTAGGGTCGATAACGTGAGGGG | 0% | 81% |
| GGGGTTGTAGGGTCGATAACGTGAGTAA | 0% | 45% |
| GGGGTTGTAGGGTCGATAACGTGAGCAA | 0% | 34% |
| GGGGTTGTAGGGTCGATAACGTGAGGAA | 0% | 45% |
| GGGGTTGTAGGGTCGATAACGTGAGTCA | 0% | 40% |
| GGGGTTGTAGGGTCGATAACGTGAGCCA | 0% | 35% |
| GGGGTTGTAGGGTCGATAACGTGAGGCA | 0% | 29% |
| GGGGTTGTAGGGTCGATAACGTGAGTGA | 0% | 40% |
| GGGGTTGTAGGGTCGATAACGTGAGCGA | 1% | 18% |
| GGGGTTGTAGGGTCGATAACGTGAGGGA | 2% | 79% |
| GGGGTTGTAGGGTCGATAACGTGAGTAT | 0% | 52% |
| GGGGTTGTAGGGTCGATAACGTGAGCAT | 0% | 45% |
| GGGGTTGTAGGGTCGATAACGTGAGGAT | 10% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGTCT | 9% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGCCT | 0% | 0% |
| GGGGTTGTAGGGTCGATAACGTGAGGCT | 0% | 100% |
| GGGGTTGTAGGGTCGATAACGTGAGTGT | 0% | 55% |
| GGGGTTGTAGGGTCGATAACGTGAGCGT | 0% | 20% |
| GGGGTTGTAGGGTCGATAACGTGAGGGT | 0% | 100% |
| GGGGTTGTAGGGTCGATAACGTGATCGA | 0% | 3% |
| GGGGTTGTAGGGTCGATAACGTGACGAA | 0% | 14% |
| GGGGTTGTAGGGTCGATAACGTGCTCGA | 0% | 0% |
| GGGGTTGTAGGGTCGATAACGTGGCGAA | 0% | 4% |
| Primers | Nucleotide Sequences (5’–3’) | Ref. | |
|---|---|---|---|
| PCR-1 | Babesia-UP * | TAGTGACAAGAAATAACAATACAGGGCG | This study |
| Theileria-UP | TAGTGACAAGAAATAACAATACGGGGCT | ||
| Ba/Th-DN | GCTTTCGCAGTAGTTCGTCTTTAACAA | ||
| PCR-2 | Babesia-UP | AATGTCTTGTAATTGGAATGATGGT | |
| Theileria-UP | AATGTCTTGTAATTGGAATGATGGG | ||
| Ba/Th-DN | TTCGCAGTAGTTCGTCTTTAACAA | ||
| PCR-3 | Ba/Th-UP | CGCAAATTACCCAATCCTGACA | |
| Babesia-DN | CAACTACGAGCTTTTTAACTGCAACAAG | ||
| Theileria-DN | CAACTACGAGCTTTTTAACTGCAACAAT | ||
| PCR-4 | Babesia-UP | AATTCCAGCTCCAATAGCGTATATTAAAC | |
| Theileria-UP | AATTCCAGCTCCAATAGCGTATATTAAAA | ||
| Ba/Th-DN | GCTTTCGCAGTAGTTCGTCTTTAACAA | ||
| PCR-5 | Babesia-UP | TTCAAGCAGACTTTTGTCTTGAATAC | |
| Theileria-UP | TCAAAGCAGGCTTTTGCCTTGAATAG | ||
| Ba/Th-DN | GCTTTCGCAGTAGTTCGTCTTTAACAA | ||
| PCR-6 | BJ1 | GTCTTGTAATTGGAATGATGG | [21] |
| BN2 | TAGTTTATGGTTAGGACTACG | ||
| PCR-7 | PIRO-A | AATACCCAATCCTGACACAGGG | [20] |
| PIRO-B | TTAAATACGAATGCCCCCAAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, K.; Zhang, J.; Li, J.; Qiu, H.; Wei, L.; Yang, Y.; Wang, C. Exploring the Impact of Primer–Template Mismatches on PCR Performance of DNA Polymerases Varying in Proofreading Activity. Genes 2024, 15, 215. https://doi.org/10.3390/genes15020215
Huang K, Zhang J, Li J, Qiu H, Wei L, Yang Y, Wang C. Exploring the Impact of Primer–Template Mismatches on PCR Performance of DNA Polymerases Varying in Proofreading Activity. Genes. 2024; 15(2):215. https://doi.org/10.3390/genes15020215
Chicago/Turabian StyleHuang, Ke, Jilei Zhang, Jing Li, Haixiang Qiu, Lanjing Wei, Yi Yang, and Chengming Wang. 2024. "Exploring the Impact of Primer–Template Mismatches on PCR Performance of DNA Polymerases Varying in Proofreading Activity" Genes 15, no. 2: 215. https://doi.org/10.3390/genes15020215
APA StyleHuang, K., Zhang, J., Li, J., Qiu, H., Wei, L., Yang, Y., & Wang, C. (2024). Exploring the Impact of Primer–Template Mismatches on PCR Performance of DNA Polymerases Varying in Proofreading Activity. Genes, 15(2), 215. https://doi.org/10.3390/genes15020215