
Chemogenetic Inhibition of Prefrontal Cortex Ameliorates Autism-Like Social Deficits and Absence-Like Seizures in a Gene-Trap Ash1l Haploinsufficiency Mouse Model


Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Care and Husbandry
2.2. Behavioral Tests
2.3. LacZ (β-galactosidase) Staining
2.4. Pentylenetetrazol (PTZ, a Competitive GABAA Receptor Antagonist) Administration
2.5. EEG
2.6. Brain Slice Preparation
2.7. Whole-Cell Patch-Clamp Recordings
2.8. Quantitative Real-Time PCR
2.9. Viral Vectors and Animal Surgery
2.10. Western Blotting
2.11. Statistical Analysis
3. Results
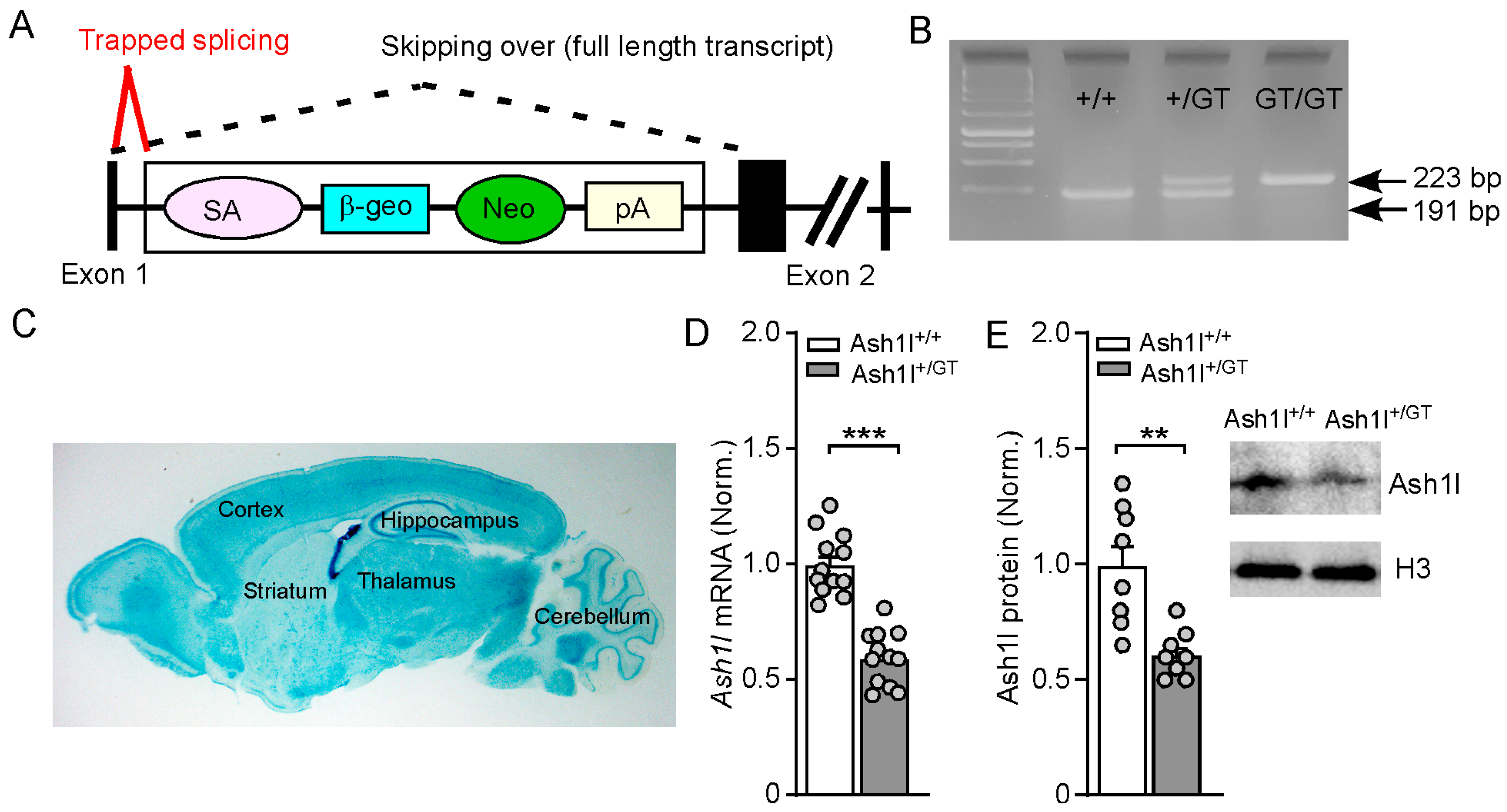
3.1. Validation of Gene-Trap Knockout of Ash1l in the Brain
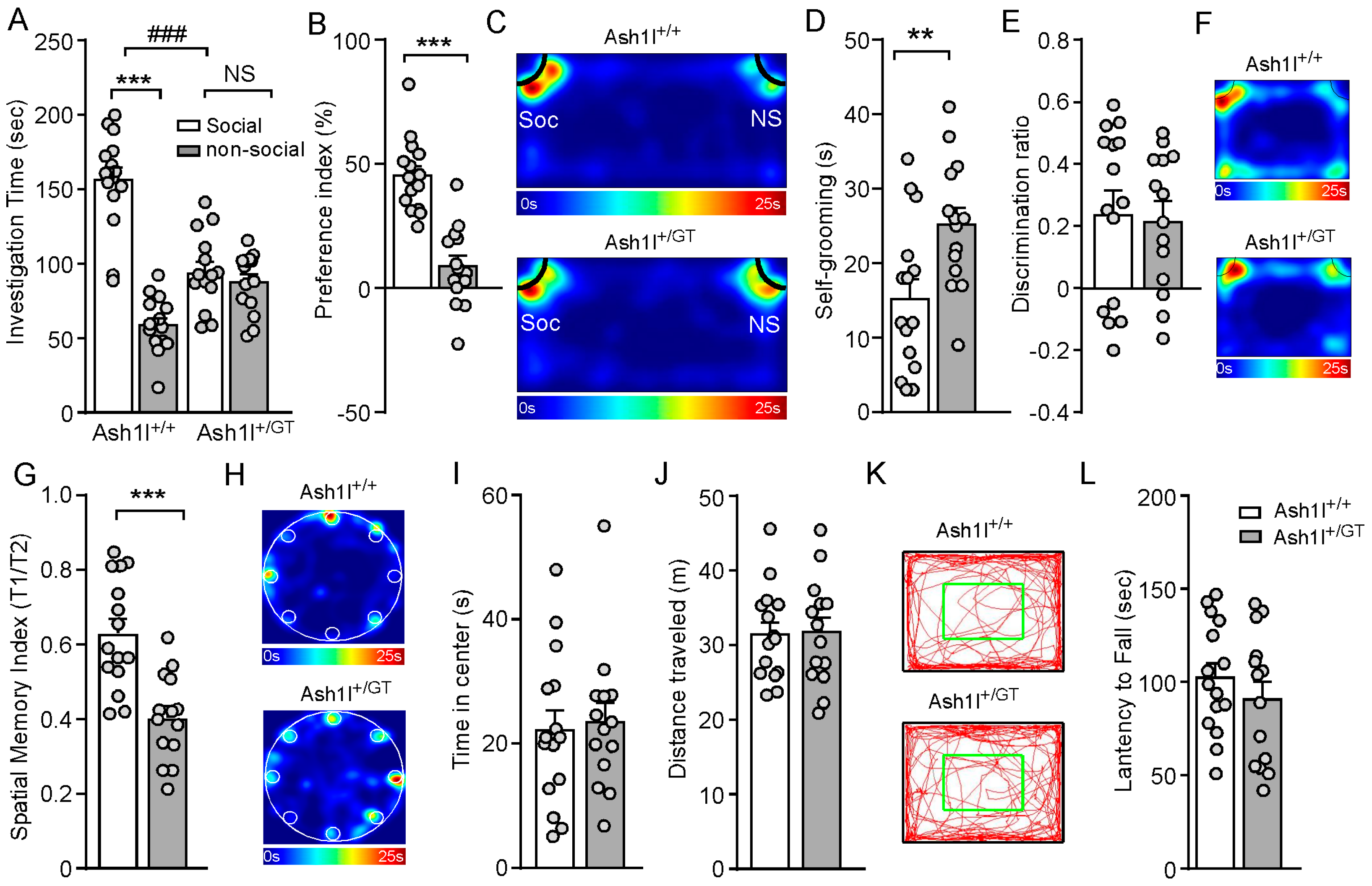
3.2. Ash1l Haploinsufficiency Causes Autism-Like Behavioral Deficits in Male and Female Mice
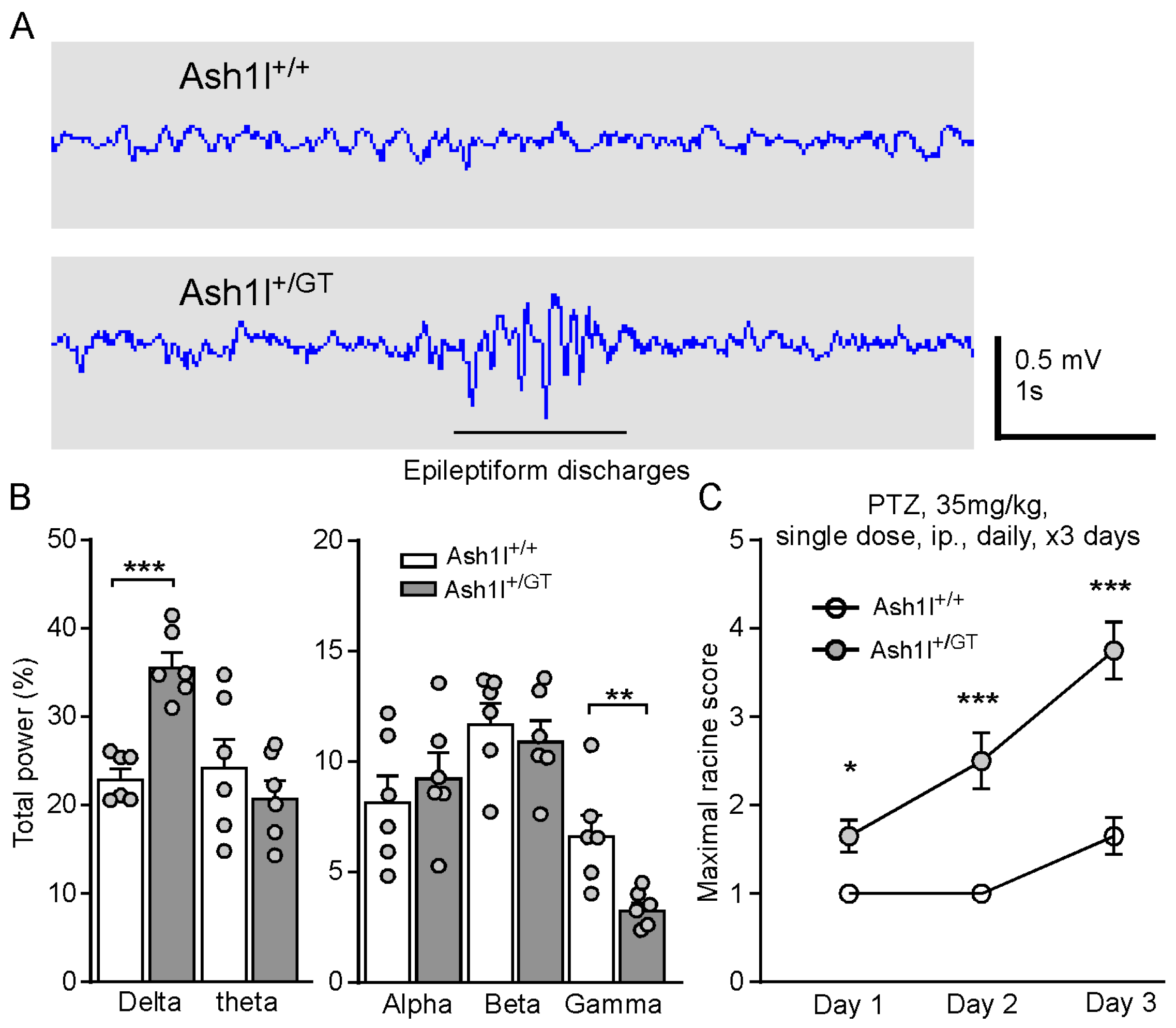
3.3. Ash1l Haploinsufficiency Causes Absence-Like Seizures and Increases the Susceptibility for Convulsive Seizures
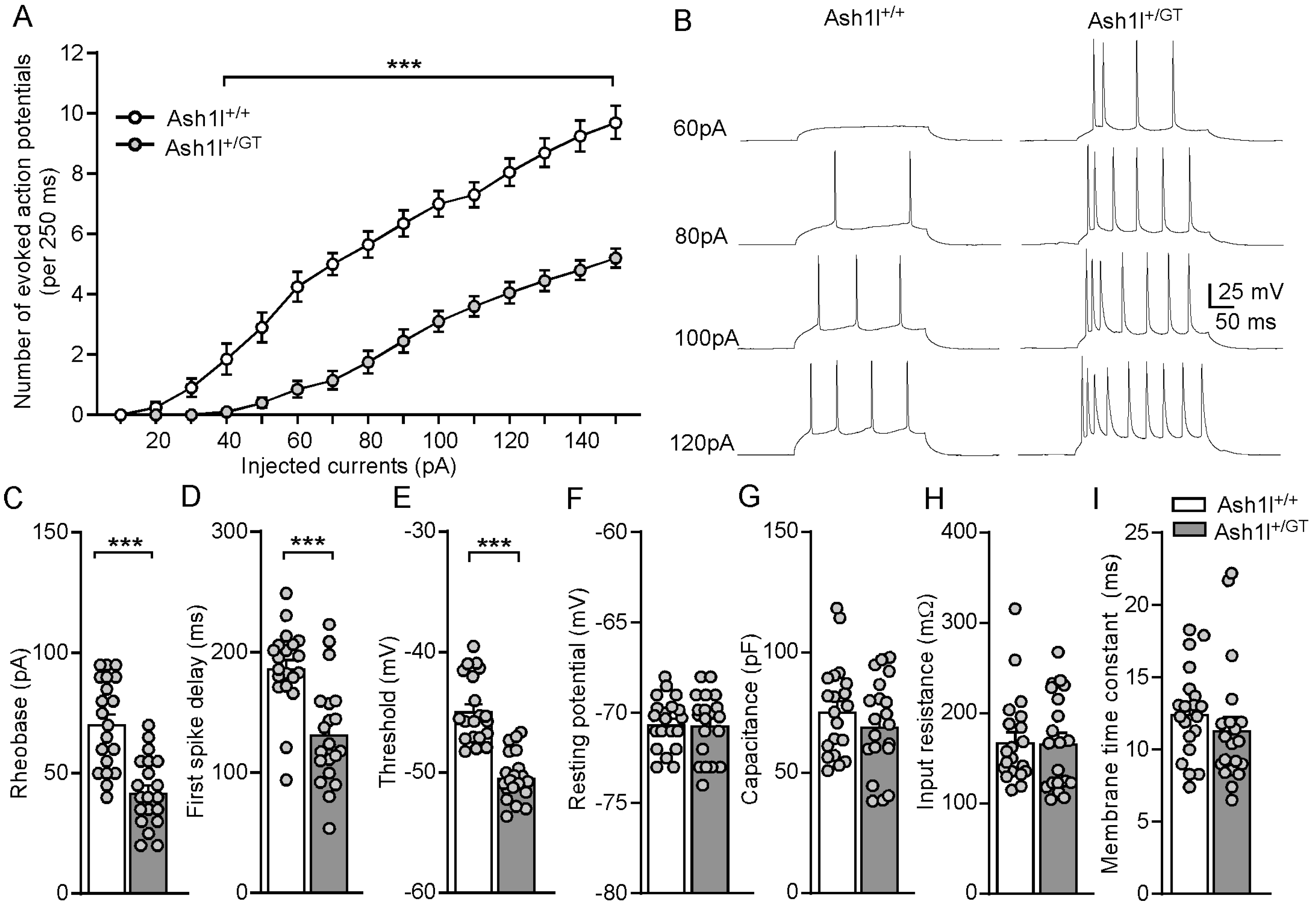
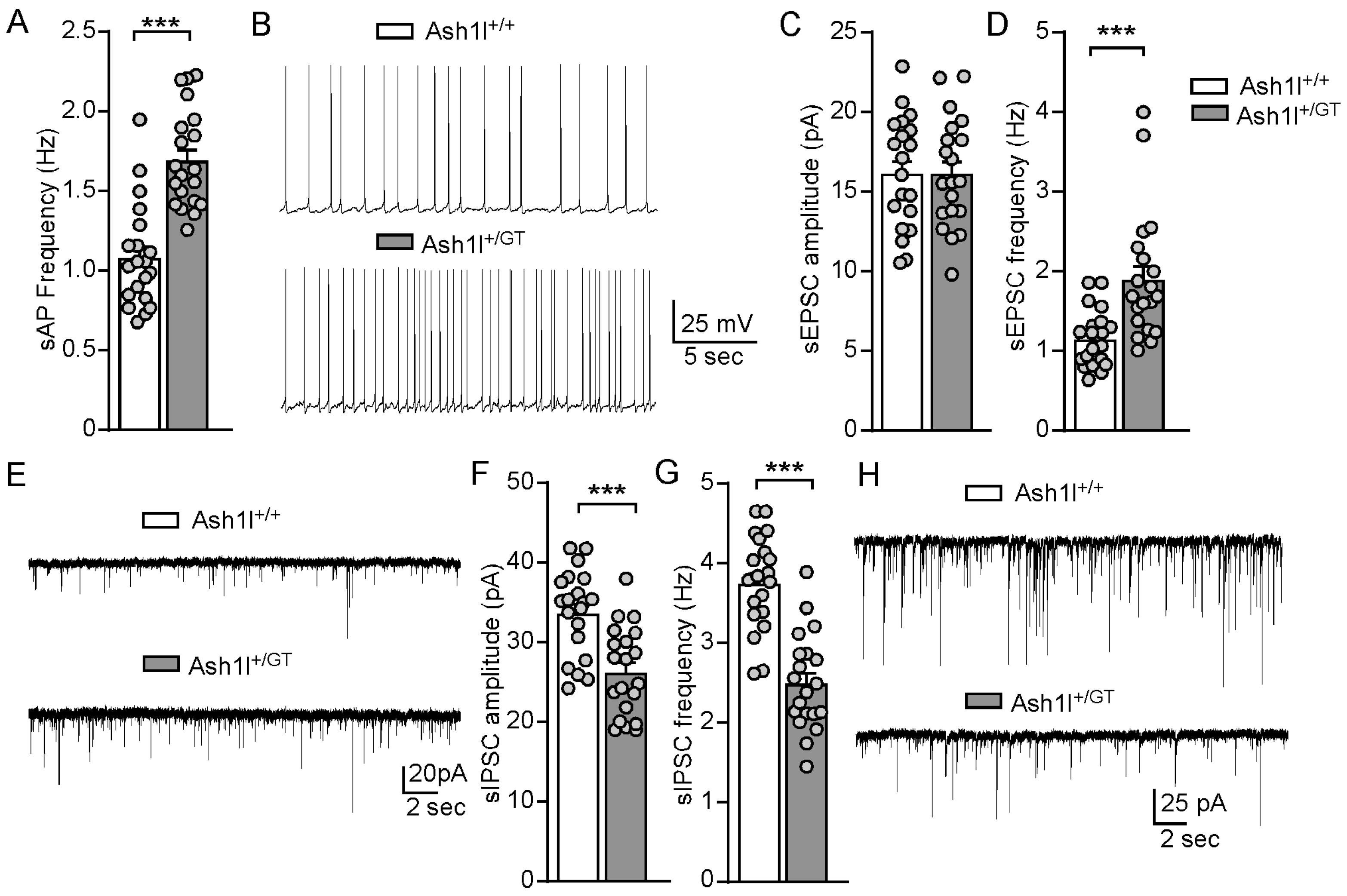
3.4. Ash1l Haploinsufficiency Increases the Excitability of Pyramidal Neurons in the PFC
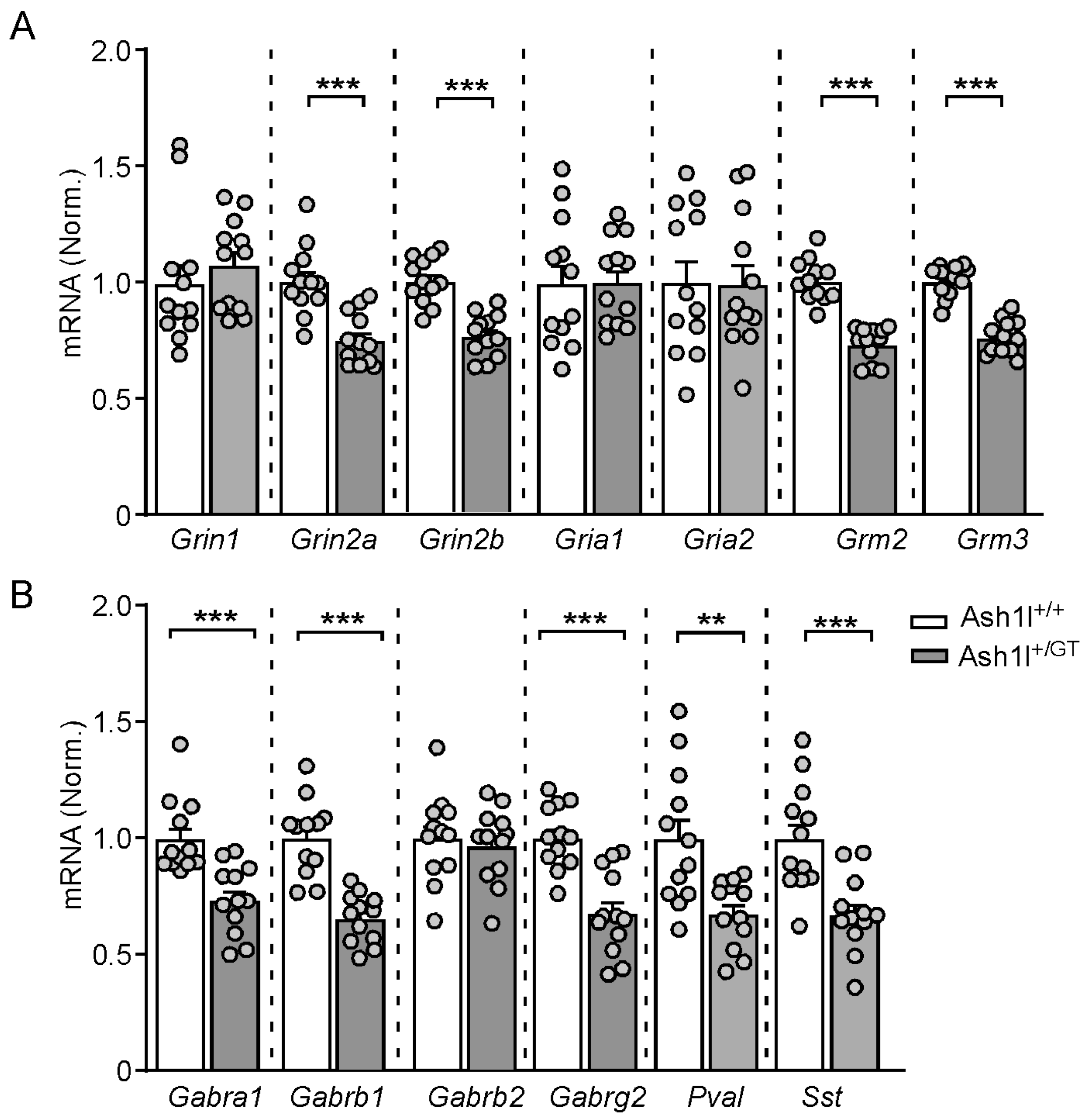
3.5. Ash1l Haploinsufficiency Alters the Transcriptional Levels of the Key Excitatory and Inhibitory Synaptic Genes in the PFC
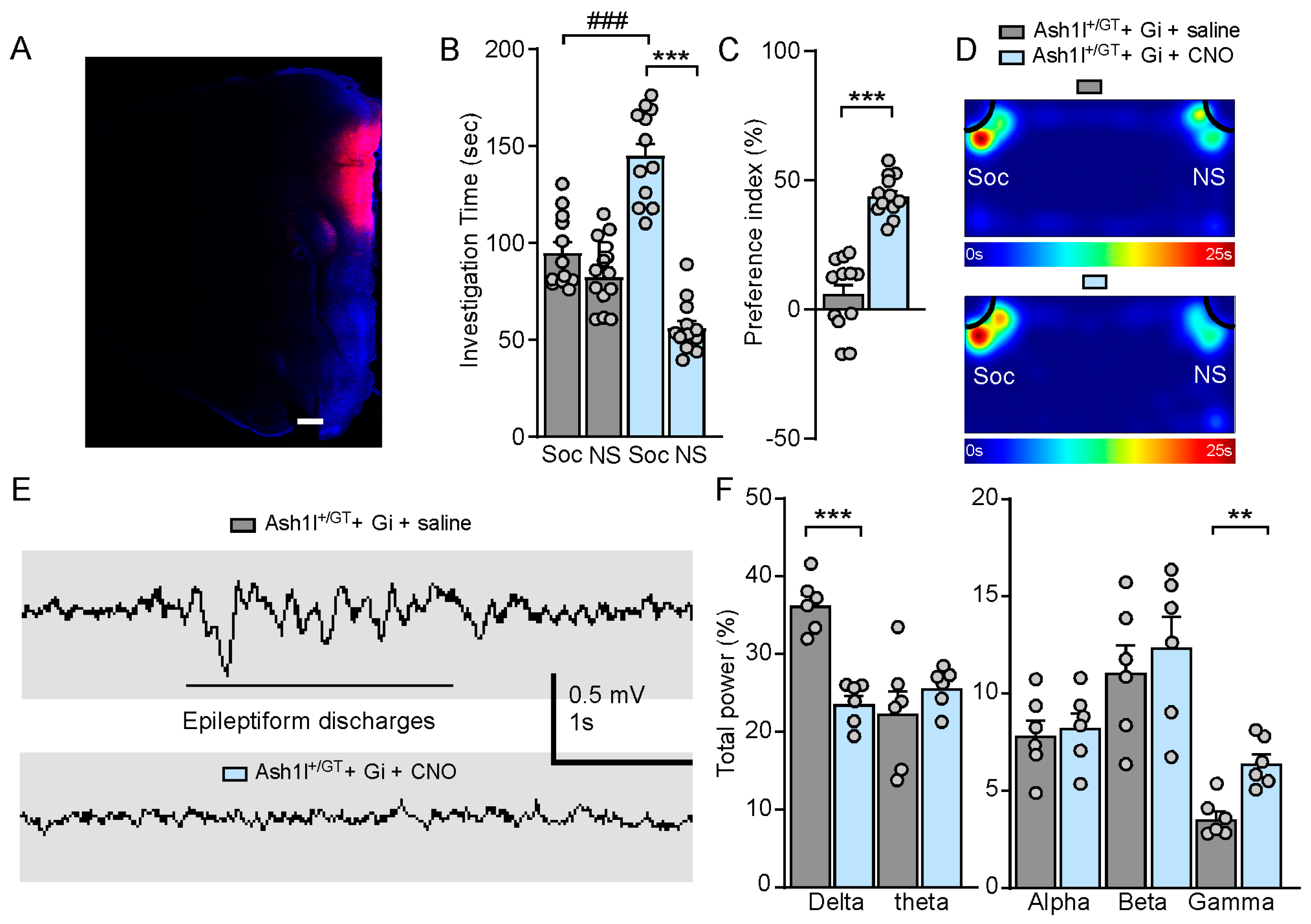
3.6. Chemogenetic Inhibition of Pyramidal Neurons in the PFC Ameliorates Autism-Like Social Deficits and Abolishes Absence-Like Seizures in Ash1l Haploinsufficiency Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blackmon, K.; Bluvstein, J.; MacAllister, W.S.; Avallone, J.; Misajon, J.; Hedlund, J.; Goldberg, R.; Bojko, A.; Mitra, N.; Giridharan, R.; et al. Treatment Resistant Epilepsy in Autism Spectrum Disorder: Increased Risk for Females. Autism Res. 2016, 9, 311–320. [Google Scholar] [CrossRef]
- Frye, R.E.; Rossignol, D.; Casanova, M.F.; Brown, G.L.; Martin, V.; Edelson, S.; Coben, R.; Lewine, J.; Slattery, J.C.; Lau, C.; et al. A review of traditional and novel treatments for seizures in autism spectrum disorder: Findings from a systematic review and expert panel. Front. Public Health 2013, 1, 31. [Google Scholar] [CrossRef]
- Morrison-Levy, N.; Go, C.; Ochi, A.; Otsubo, H.; Drake, J.; Rutka, J.; Weiss, S.K. Children with autism spectrum disorders and drug-resistant epilepsy can benefit from epilepsy surgery. Epilepsy Behav. 2018, 85, 200–204. [Google Scholar] [CrossRef] [PubMed]
- Hyman, S.L.; Levy, S.E.; Myers, S.M. Identification, Evaluation, and Management of Children With Autism Spectrum Disorder. Pediatrics 2020, 145, e20193447. [Google Scholar] [CrossRef] [PubMed]
- Viscidi, E.W.; Triche, E.W.; Pescosolido, M.F.; McLean, R.L.; Joseph, R.M.; Spence, S.J.; Morrow, E.M. Clinical characteristics of children with autism spectrum disorder and co-occurring epilepsy. PLoS ONE 2013, 8, e67797. [Google Scholar] [CrossRef] [PubMed]
- Jeste, S.S.; Tuchman, R. Autism Spectrum Disorder and Epilepsy: Two Sides of the Same Coin? J. Child. Neurol. 2015, 30, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e523. [Google Scholar] [CrossRef]
- Stessman, H.A.; Xiong, B.; Coe, B.P.; Wang, T.; Hoekzema, K.; Fenckova, M.; Kvarnung, M.; Gerdts, J.; Trinh, S.; Cosemans, N.; et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017, 49, 515–526. [Google Scholar] [CrossRef]
- Iossifov, I.; Levy, D.; Allen, J.; Ye, K.; Ronemus, M.; Lee, Y.H.; Yamrom, B.; Wigler, M. Low load for disruptive mutations in autism genes and their biased transmission. Proc. Natl. Acad. Sci. USA 2015, 112, E5600–E5607. [Google Scholar] [CrossRef] [PubMed]
- Tammimies, K.; Marshall, C.R.; Walker, S.; Kaur, G.; Thiruvahindrapuram, B.; Lionel, A.C.; Yuen, R.K.; Uddin, M.; Roberts, W.; Weksberg, R.; et al. Molecular Diagnostic Yield of Chromosomal Microarray Analysis and Whole-Exome Sequencing in Children With Autism Spectrum Disorder. JAMA 2015, 314, 895–903. [Google Scholar] [CrossRef]
- Okamoto, N.; Miya, F.; Tsunoda, T.; Kato, M.; Saitoh, S.; Yamasaki, M.; Kanemura, Y.; Kosaki, K. Novel MCA/ID syndrome with ASH1L mutation. Am. J. Med. Genet. A 2017, 173, 1644–1648. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Guo, H.; Xiong, B.; Stessman, H.A.; Wu, H.; Coe, B.P.; Turner, T.N.; Liu, Y.; Zhao, W.; Hoekzema, K.; et al. De novo genic mutations among a Chinese autism spectrum disorder cohort. Nat. Commun. 2016, 7, 13316. [Google Scholar] [CrossRef] [PubMed]
- de Ligt, J.; Willemsen, M.H.; van Bon, B.W.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C.; et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Tian, M.; He, F.; Li, J.; Xie, H.; Liu, W.; Zhang, Y.; Zhang, R.; Yi, M.; Che, F.; et al. Mutations in ASH1L confer susceptibility to Tourette syndrome. Mol. Psychiatry 2020, 25, 476–490. [Google Scholar] [CrossRef]
- Yan, Y.; Tian, M.; Li, M.; Zhou, G.; Chen, Q.; Xu, M.; Hu, Y.; Luo, W.; Guo, X.; Zhang, C.; et al. ASH1L haploinsufficiency results in autistic-like phenotypes in mice and links Eph receptor gene to autism spectrum disorder. Neuron 2022, 110, 1156–1172.e9. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Duque-Wilckens, N.; Aljazi, M.B.; Wu, Y.; Moeser, A.J.; Mias, G.I.; Robison, A.J.; He, J. Loss of histone methyltransferase ASH1L in the developing mouse brain causes autistic-like behaviors. Commun. Biol. 2021, 4, 756. [Google Scholar] [CrossRef]
- Qin, L.; Williams, J.B.; Tan, T.; Liu, T.; Cao, Q.; Ma, K.; Yan, Z. Deficiency of autism risk factor ASH1L in prefrontal cortex induces epigenetic aberrations and seizures. Nat. Commun. 2021, 12, 6589. [Google Scholar] [CrossRef]
- Lee, E.; Lee, J.; Kim, E. Excitation/Inhibition Imbalance in Animal Models of Autism Spectrum Disorders. Biol. Psychiatry 2016, 81, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Penzes, P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med. 2015, 15, 146–167. [Google Scholar] [CrossRef] [PubMed]
- Masuda, F.; Nakajima, S.; Miyazaki, T.; Yoshida, K.; Tsugawa, S.; Wada, M.; Ogyu, K.; Croarkin, P.E.; Blumberger, D.M.; Daskalakis, Z.J.; et al. Motor cortex excitability and inhibitory imbalance in autism spectrum disorder assessed with transcranial magnetic stimulation: A systematic review. Transl. Psychiatry 2019, 9, 110. [Google Scholar] [CrossRef] [PubMed]
- Ziburkus, J.; Cressman, J.R.; Schiff, S.J. Seizures as imbalanced up states: Excitatory and inhibitory conductances during seizure-like events. J. Neurophysiol. 2013, 109, 1296–1306. [Google Scholar] [CrossRef] [PubMed]
- Yizhar, O.; Fenno, L.E.; Prigge, M.; Schneider, F.; Davidson, T.J.; O’Shea, D.J.; Sohal, V.S.; Goshen, I.; Finkelstein, J.; Paz, J.T.; et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 2011, 477, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Addis, L.; Virdee, J.K.; Vidler, L.R.; Collier, D.A.; Pal, D.K.; Ursu, D. Epilepsy-associated GRIN2A mutations reduce NMDA receptor trafficking and agonist potency—Molecular profiling and functional rescue. Sci. Rep. 2017, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Oyrer, J.; Maljevic, S.; Scheffer, I.E.; Berkovic, S.F.; Petrou, S.; Reid, C.A. Ion Channels in Genetic Epilepsy: From Genes and Mechanisms to Disease-Targeted Therapies. Pharmacol. Rev. 2018, 70, 142–173. [Google Scholar] [CrossRef]
- Spratt, P.W.E.; Ben-Shalom, R.; Keeshen, C.M.; Burke, K.J., Jr.; Clarkson, R.L.; Sanders, S.J.; Bender, K.J. The Autism-Associated Gene Scn2a Contributes to Dendritic Excitability and Synaptic Function in the Prefrontal Cortex. Neuron 2019, 103, 673–685.e5. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, X.; Eaton, M.; Wu, J.; Ma, Z.; Lai, S.; Park, A.; Ahmad, T.S.; Que, Z.; Lee, J.H.; et al. Severe deficiency of the voltage-gated sodium channel Na(V)1.2 elevates neuronal excitability in adult mice. Cell Rep. 2021, 36, 109495. [Google Scholar] [CrossRef]
- Qin, L.; Ma, K.; Yan, Z. Chemogenetic Activation of Prefrontal Cortex in Shank3-Deficient Mice Ameliorates Social Deficits, NMDAR Hypofunction, and Sgk2 Downregulation. iScience 2019, 17, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Ma, K.; Wang, Z.J.; Hu, Z.; Matas, E.; Wei, J.; Yan, Z. Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nat. Neurosci. 2018, 21, 564–575. [Google Scholar] [CrossRef]
- Sacai, H.; Sakoori, K.; Konno, K.; Nagahama, K.; Suzuki, H.; Watanabe, T.; Watanabe, M.; Uesaka, N.; Kano, M. Autism spectrum disorder-like behavior caused by reduced excitatory synaptic transmission in pyramidal neurons of mouse prefrontal cortex. Nat. Commun. 2020, 11, 5140. [Google Scholar] [CrossRef] [PubMed]
- Chini, M.; Hanganu-Opatz, I.L. Prefrontal Cortex Development in Health and Disease: Lessons from Rodents and Humans. Trends Neurosci. 2021, 44, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Somel, M.; Liu, X.; Khaitovich, P. Human brain evolution: Transcripts, metabolites and their regulators. Nat. Rev. Neurosci. 2013, 14, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Forrest, M.P.; Parnell, E.; Penzes, P. Dendritic structural plasticity and neuropsychiatric disease. Nat. Rev. Neurosci. 2018, 19, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Somel, M.; Tang, L.; Yan, Z.; Jiang, X.; Guo, S.; Yuan, Y.; He, L.; Oleksiak, A.; Zhang, Y.; et al. Extension of cortical synaptic development distinguishes humans from chimpanzees and macaques. Genome Res. 2012, 22, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Brinkmeier, M.L.; Geister, K.A.; Jones, M.; Waqas, M.; Maillard, I.; Camper, S.A. The Histone Methyltransferase Gene Absent, Small, or Homeotic Discs-1 Like Is Required for Normal Hox Gene Expression and Fertility in Mice. Biol. Reprod. 2015, 93, 121. [Google Scholar] [CrossRef]
- Rogan, S.C.; Roth, B.L. Remote control of neuronal signaling. Pharmacol. Rev. 2011, 63, 291–315. [Google Scholar] [CrossRef]
- Roth, B.L. DREADDs for Neuroscientists. Neuron 2016, 89, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Taylor, C.; Williamson, M.; Newton, S.S.; Qin, L. Diminished activity-dependent BDNF signaling differentially causes autism-like behavioral deficits in male and female mice. Front. Psychiatry 2023, 14, 1182472. [Google Scholar] [CrossRef]
- Sharma, S.; Puttachary, S.; Thippeswamy, A.; Kanthasamy, A.G.; Thippeswamy, T. Status Epilepticus: Behavioral and Electroencephalography Seizure Correlates in Kainate Experimental Models. Front. Neurol. 2018, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Racine, R.J. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Shimada, T.; Yamagata, K. Pentylenetetrazole-Induced Kindling Mouse Model. J. Vis. Exp. JoVE 2018, 12, 56573. [Google Scholar] [CrossRef]
- Tan, T.; Wang, W.; Liu, T.; Zhong, P.; Conrow-Graham, M.; Tian, X.; Yan, Z. Neural circuits and activity dynamics underlying sex-specific effects of chronic social isolation stress. Cell Rep. 2021, 34, 108874. [Google Scholar] [CrossRef]
- Sun, Q.; Jiang, Y.Q.; Lu, M.C. Topographic heterogeneity of intrinsic excitability in mouse hippocampal CA3 pyramidal neurons. J. Neurophysiol. 2020, 124, 1270–1284. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Zhang, D.; McDaniel, K.; Webb, M.; Newton, S.S.; Lee, F.S.; Qin, L. A sexually dimorphic signature of activity-dependent BDNF signaling on the intrinsic excitability of pyramidal neurons in the prefrontal cortex. Front. Cell. Neurosci. 2024, 18, 1496930. [Google Scholar] [CrossRef] [PubMed]
- Kalueff, A.V.; Stewart, A.M.; Song, C.; Berridge, K.C.; Graybiel, A.M.; Fentress, J.C. Neurobiology of rodent self-grooming and its value for translational neuroscience. Nat. Rev. Neurosci. 2016, 17, 45–59. [Google Scholar] [CrossRef]
- Pitts, M.W. Barnes Maze Procedure for Spatial Learning and Memory in Mice. Bio Protoc. 2018, 8, e2744. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cao, Q.; Tan, T.; Yang, F.; Williams, J.B.; Yan, Z. Epigenetic treatment of behavioral and physiological deficits in a tauopathy mouse model. Aging Cell 2021, 20, e13456. [Google Scholar] [CrossRef] [PubMed]
- Stoner, R.; Chow, M.L.; Boyle, M.P.; Sunkin, S.M.; Mouton, P.R.; Roy, S.; Wynshaw-Boris, A.; Colamarino, S.A.; Lein, E.S.; Courchesne, E. Patches of disorganization in the neocortex of children with autism. N. Engl. J. Med. 2014, 370, 1209–1219. [Google Scholar] [CrossRef]
- Chen, L.; Cummings, K.A.; Mau, W.; Zaki, Y.; Dong, Z.; Rabinowitz, S.; Clem, R.L.; Shuman, T.; Cai, D.J. The role of intrinsic excitability in the evolution of memory: Significance in memory allocation, consolidation, and updating. Neurobiol. Learn. Mem. 2020, 173, 107266. [Google Scholar] [CrossRef]
- Armbruster, B.N.; Li, X.; Pausch, M.H.; Herlitze, S.; Roth, B.L. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. USA 2007, 104, 5163–5168. [Google Scholar] [CrossRef]
- Silverman, J.L.; Yang, M.; Lord, C.; Crawley, J.N. Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 2010, 11, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Faundes, V.; Newman, W.G.; Bernardini, L.; Canham, N.; Clayton-Smith, J.; Dallapiccola, B.; Davies, S.J.; Demos, M.K.; Goldman, A.; Gill, H.; et al. Histone Lysine Methylases and Demethylases in the Landscape of Human Developmental Disorders. Am. J. Hum. Genet. 2018, 102, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Cordova, I.; Blesson, A.; Savatt, J.M.; Sveden, A.; Mahida, S.; Hazlett, H.; Rooney Riggs, E.; Chopra, M. Expansion of the Genotypic and Phenotypic Spectrum of ASH1L-Related Syndromic Neurodevelopmental Disorder. Genes 2024, 15, 423. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, D.T.; Lan, S.; Yang, Y.; Huang, J.; Huang, J.; Fang, L. ASH1L mutation caused seizures and intellectual disability in twin sisters. J. Clin. Neurosci. 2021, 91, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Siniscalco, D.; Cirillo, A.; Bradstreet, J.J.; Antonucci, N. Epigenetic findings in autism: New perspectives for therapy. Int. J. Environ. Res. Public Health 2013, 10, 4261–4273. [Google Scholar] [CrossRef]
- Shulha, H.P.; Cheung, I.; Whittle, C.; Wang, J.; Virgil, D.; Lin, C.L.; Guo, Y.; Lessard, A.; Akbarian, S.; Weng, Z. Epigenetic signatures of autism: Trimethylated H3K4 landscapes in prefrontal neurons. Arch. Gen. Psychiatry 2012, 69, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Cheung, I.; Shulha, H.P.; Jiang, Y.; Matevossian, A.; Wang, J.; Weng, Z.; Akbarian, S. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc. Natl. Acad. Sci. USA 2010, 107, 8824–8829. [Google Scholar] [CrossRef]
- Gregory, G.D.; Vakoc, C.R.; Rozovskaia, T.; Zheng, X.; Patel, S.; Nakamura, T.; Canaani, E.; Blobel, G.A. Mammalian ASH1L is a histone methyltransferase that occupies the transcribed region of active genes. Mol. Cell. Biol. 2007, 27, 8466–8479. [Google Scholar] [CrossRef] [PubMed]
- De, I.; Muller, C.W. Unleashing the Power of ASH1L Methyltransferase. Structure 2019, 27, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Ma, K.; Yan, Z. Rescue of histone hypoacetylation and social deficits by ketogenic diet in a Shank3 mouse model of autism. Neuropsychopharmacology 2021, 47, 1271–1279. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Forward | Reverse | Gene Reference | Length (bp) |
|---|---|---|---|---|
| Gapdh | gacaactcactcaagattgtcag | atggcatggactgtggtcatgag | NM_001289726.1 | 122 |
| Ash1l | tgggaagatgacagatgaga | aagatggatgctttcttcgg | NM_138679.5 | 121 |
| Grin1 | catcggacttcagctaatca | gtccccatcctcattgaatt | NM_008169.3 | 238 |
| Grin2a | ggctacagagacttcatcag | atccagaagaaatcgtagcc | NM_008170.4 | 233 |
| Grin2b | ttaacaactccgtacctgtg | tggaacttcttgtcactcag | NM_008171.4 | 175 |
| Gria1 | gccttaatcgagttctgcta | gaatggattgcatggacttg | NM_008165.4 | 205 |
| Gria2 | agcctatgagatctggatgt | gagagagatcttggcgaaat | NM_001083806.3 | 228 |
| Grm2 | gcttaggttcctggcact | ttaacaggtccacactcctc | NM_001160353 | 150 |
| Grm3 | caattacttgcttccaggag | tagtcaacgatgctctgaca | NM_181850 | 110 |
| Gabra1 | caccatgaggttgaccgtga | ctacaaccactgaacgggct | NM_010250.5 | 158 |
| Gabrb1 | catagacatggtctcggaag | gtcagctactctgttgtcaa | NM_008069 | 130 |
| Gabrb2 | atttggtggctcaaacggtc | gagatttcctcaccagcagga | NM_008070.4 | 168 |
| Gabrg2 | ggagccggcatcaaatcatc | cttttggcttgtgaagcctgg | NM_008073.4 | 214 |
| Pvalb | ggtgaagaaggtgttccata | cagacaagtctctggcatct | NM_001330686 | 110 |
| Sst | cagactccgtcagtttctgc | atcattctctgtctggttgg | NM_009215.1 | 112 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, K.; McDaniel, K.; Zhang, D.; Webb, M.; Qin, L. Chemogenetic Inhibition of Prefrontal Cortex Ameliorates Autism-Like Social Deficits and Absence-Like Seizures in a Gene-Trap Ash1l Haploinsufficiency Mouse Model. Genes 2024, 15, 1619. https://doi.org/10.3390/genes15121619
Ma K, McDaniel K, Zhang D, Webb M, Qin L. Chemogenetic Inhibition of Prefrontal Cortex Ameliorates Autism-Like Social Deficits and Absence-Like Seizures in a Gene-Trap Ash1l Haploinsufficiency Mouse Model. Genes. 2024; 15(12):1619. https://doi.org/10.3390/genes15121619
Chicago/Turabian StyleMa, Kaijie, Kylee McDaniel, Daoqi Zhang, Maria Webb, and Luye Qin. 2024. "Chemogenetic Inhibition of Prefrontal Cortex Ameliorates Autism-Like Social Deficits and Absence-Like Seizures in a Gene-Trap Ash1l Haploinsufficiency Mouse Model" Genes 15, no. 12: 1619. https://doi.org/10.3390/genes15121619
APA StyleMa, K., McDaniel, K., Zhang, D., Webb, M., & Qin, L. (2024). Chemogenetic Inhibition of Prefrontal Cortex Ameliorates Autism-Like Social Deficits and Absence-Like Seizures in a Gene-Trap Ash1l Haploinsufficiency Mouse Model. Genes, 15(12), 1619. https://doi.org/10.3390/genes15121619