
Exploring the Role of CCNF Variants in Italian ALS Patients
 , ,
, ,  ,
,  , and
, and
Abstract
1. Introduction
2. Patients and Methods
Genetic Analysis
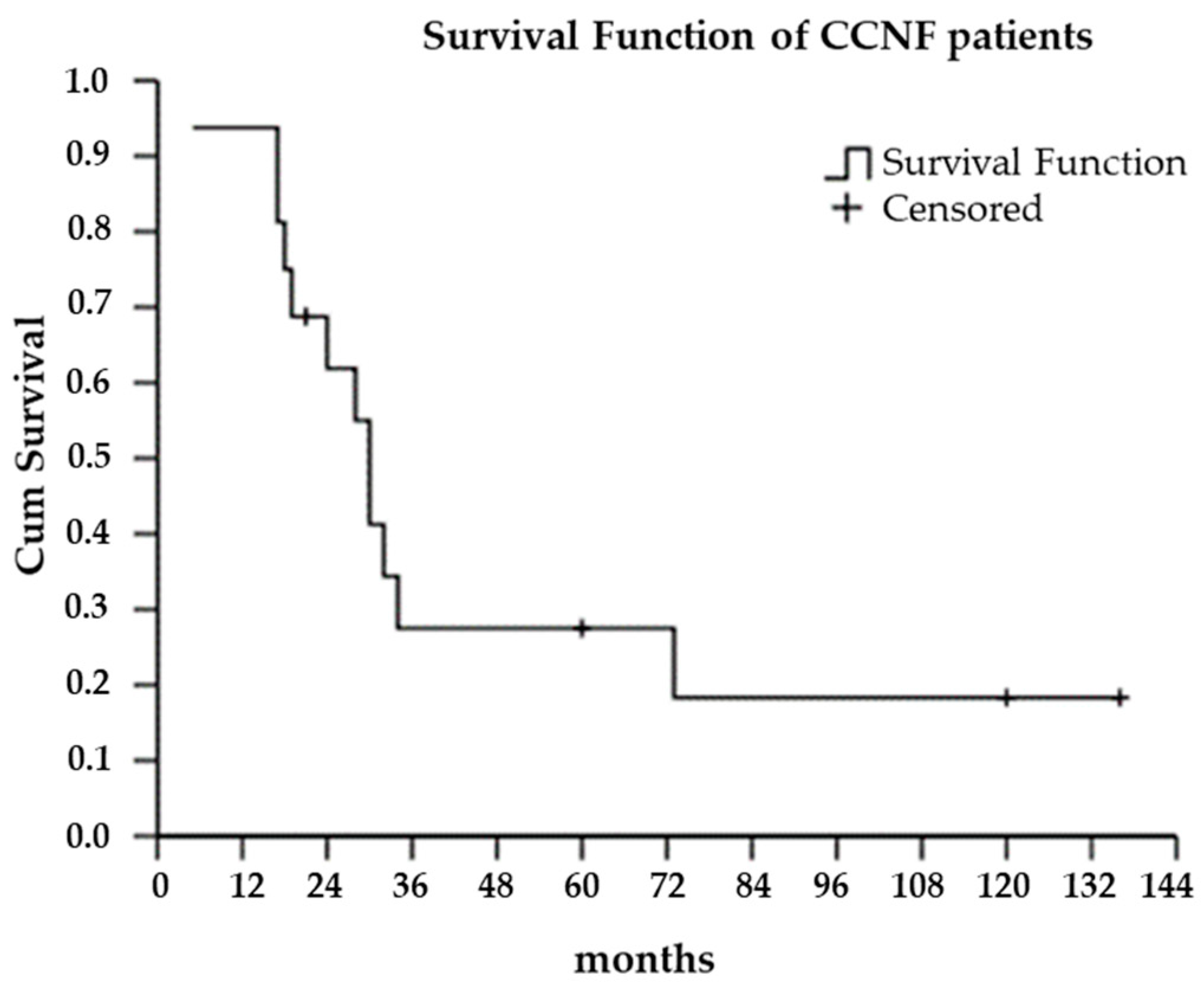
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brown, R.H., Jr.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 1602. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Williams, K.L.; Topp, S.; Yang, S.; Smith, B.; Fifita, J.A.; Warraich, S.T.; Zhang, K.Y.; Farrawell, N.; Vance, C.; Hu, X.; et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun. 2016, 7, 11253. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bai, C.; Richman, R.; Elledge, S.J. Human cyclin F. EMBO J. 1994, 13, 6087–6098. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Galper, J.; Rayner, S.L.; Hogan, A.L.; Fifita, J.A.; Lee, A.; Chung, R.S.; Blair, I.P.; Yang, S. Cyclin F: A component of an E3 ubiquitin ligase complex with roles in neurodegeneration and cancer. Int. J. Biochem. Cell Biol. 2017, 89, 216–220. [Google Scholar] [CrossRef] [PubMed]
- D’Angiolella, V.; Esencay, M.; Pagano, M. A cyclin without cyclin-dependent kinases: Cyclin F controls genome stability through ubiquitin-mediated proteolysis. Trends Cell Biol. 2013, 23, 135–140. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ragagnin, A.M.G.; Sundaramoorthy, V.; Farzana, F.; Gautam, S.; Saravanabavan, S.; Takalloo, Z.; Mehta, P.; Do-Ha, D.; Parakh, S.; Shadfar, S.; et al. ALS/FTD-associated mutation in cyclin F inhibits ER-Golgi trafficking, inducing ER stress, ERAD and Golgi fragmentation. Sci. Rep. 2023, 13, 20467. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brooks, B.R.; Miller, R.G.; Swash, M.; Munsat, T.L.; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: Revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Mot. Neuron. Disord. 2000, 1, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Sabatelli, M.; Conte, A.; Zollino, M. Clinical and genetic heterogeneity of amyotrophic lateral sclerosis. Clin. Genet. 2013, 83, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Poletti, B.; Solca, F.; Carelli, L.; Madotto, F.; Lafronza, A.; Faini, A.; Monti, A.; Zago, S.; Calini, D.; Tiloca, C.; et al. The validation of the Italian Edinburgh Cognitive and Behavioural ALS Screen (ECAS). Amyotroph. Lateral Scler. Front. Degener. 2016, 17, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; Abrahams, S.; Goldstein, L.H.; Woolley, S.; Mclaughlin, P.; Snowden, J.; Mioshi, E.; Roberts-South, A.; Benatar, M.; HortobáGyi, T.; et al. Amyotrophic lateral sclerosis-frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 153–174. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, B.; Jiang, Q.; Lin, J.; Wei, Q.; Li, C.; Hou, Y.; Cao, B.; Zhang, L.; Ou, R.; Liu, K.; et al. Genetic and Phenotypic Spectrum of Amyotrophic Lateral Sclerosis Patients with CCNF Variants from a Large Chinese Cohort. Mol. Neurobiol. 2023, 60, 4150–4160. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- McCann, E.P.; Williams, K.L.; Fifita, J.A.; Tarr, I.S.; O’Connor, J.; Rowe, D.B.; Nicholson, G.A.; Blair, I.P. The genotype-phenotype landscape of familial amyotrophic lateral sclerosis in Australia. Clin. Genet. 2017, 92, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Tripolszki, K.; Gampawar, P.; Schmidt, H.; Nagy, Z.F.; Nagy, D.; Klivényi, P.; Engelhardt, J.I.; Széll, M. Comprehensive Genetic Analysis of a Hungarian Amyotrophic Lateral Sclerosis Cohort. Front. Genet. 2019, 10, 732. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pan, C.; Jiao, B.; Xiao, T.; Hou, L.; Zhang, W.; Liu, X.; Xu, J.; Tang, B.; Shen, L. Mutations of CCNF gene is rare in patients with amyotrophic lateral sclerosis and frontotemporal dementia from Mainland China. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Li, J.; Tang, L.; Zhang, N.; Fan, D. Screening for CCNF Mutations in a Chinese Amyotrophic Lateral Sclerosis Cohort. Front. Aging Neurosci. 2018, 10, 185. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tsai, P.C.; Liao, Y.C.; Chen, P.L.; Guo, Y.C.; Chen, Y.H.; Jih, K.Y.; Lin, K.P.; Soong, B.W.; Tsai, C.P.; Lee, Y.C. Investigating CCNF mutations in a Taiwanese cohort with amyotrophic lateral sclerosis. Neurobiol. Aging 2018, 62, e1–e243. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Patient ID | Nucleotidic Change | Aminoacidic Change | Reference | gnomAD Exomes Global Allele Frequency | Additional Variants | Disease Duration (Months) | Outcome | Phenotype | SALS/ FALS | Cognitive/ Behavioural Impairment |
|---|---|---|---|---|---|---|---|---|---|---|
| 833 | c.261C > A | p.Asn87Lys | - | 0.0000206 | 136 | Alive | Flail arm | SALS | No | |
| 2496 | c.261C > A | p.Asn87Lys | - | 0.0000206 | 21 | Lost at follow-up | Flail arm | SALS | N/A | |
| 2269 | c.467C > T | p.Pro156Leu | - | 0.00000891 | 17 | Deceased | Classic | SALS | Yes (ALSci) | |
| 2354 | c.591C > A | p.Phe197Leu | - | 0.0000193 | 60 | Alive | Flail arm | FALS (possible) | No | |
| 1897 | c.591C > A | p.Phe197Leu | - | 0.0000193 | NEK1 p.Gln380Glu | 28 | Deceased | Classic | SALS | No |
| anv-1842 | c.591C > A | p.Phe197Leu | - | 0.0000193 | 120 | Alive | UMN-D | SALS | No | |
| 2097 | c.656T > C | p.Leu219Pro | - | 0.0000165 | 17 | Deceased | Classic | SALS | N/A | |
| 1713 | c.697G > C | p.Asp233His | - | 0.0000317 | 73 | Deceased | UMN-D | SALS | Yes (ALS-FTD) | |
| 2532 | c.920A > G | p.Asp307Gly | - | 0 | 19 | Deceased | Flail arm | SALS | No | |
| deg-2712 | c.1198G > A | p.Ala400Thr | - | 0.00000684 | 30 | Deceased | UMN-D | SALS | Yes (ALSbi) | |
| 2233 | c.1433C > T | p.Thr478Ile | - | 0.000000712 | 34 | Deceased | UMN-D | SALS | No | |
| 2032 | c.1525T > C | p.Ser509Pro | 4 | 0.00000616 | 24 | Deceased | Classic | SALS | No | |
| 1684 | c.1693C > A | p.Pro565Thr | - | 0.0000445 | C9orf72 expansion | 30 | Deceased | Classic | FALS (definite) | Yes (ALS-FTD) |
| 1901 | c.1697C > T | p.Ser566Leu | - | 0.0000164 | 18 | Deceased | Classic | SALS | Yes (ALSci) | |
| 1795 | c.1973C > T | p.Ala658Val | - | 0.0000158 | 5 | Tracheostomy (deceased after 29 m) | Classic | SALS | N/A | |
| 1741 | c.2194A > G | p.Ser732Gly | - | 0.0000041 | 32 | Deceased | Classic | SALS | Yes (ALS-FTD) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bisogni, G.; Conte, A.; Costantino, U.; Lattante, S.; Bernardo, D.; Lucioli, G.; Patanella, A.K.; Cimbolli, P.; Del Giudice, E.; Vettor, F.; et al. Exploring the Role of CCNF Variants in Italian ALS Patients. Genes 2024, 15, 1566. https://doi.org/10.3390/genes15121566
Bisogni G, Conte A, Costantino U, Lattante S, Bernardo D, Lucioli G, Patanella AK, Cimbolli P, Del Giudice E, Vettor F, et al. Exploring the Role of CCNF Variants in Italian ALS Patients. Genes. 2024; 15(12):1566. https://doi.org/10.3390/genes15121566
Chicago/Turabian StyleBisogni, Giulia, Amelia Conte, Umberto Costantino, Serena Lattante, Daniela Bernardo, Gabriele Lucioli, Agata Katia Patanella, Paola Cimbolli, Elda Del Giudice, Federica Vettor, and et al. 2024. "Exploring the Role of CCNF Variants in Italian ALS Patients" Genes 15, no. 12: 1566. https://doi.org/10.3390/genes15121566
APA StyleBisogni, G., Conte, A., Costantino, U., Lattante, S., Bernardo, D., Lucioli, G., Patanella, A. K., Cimbolli, P., Del Giudice, E., Vettor, F., Marangi, G., Doronzio, P. N., Zollino, M., & Sabatelli, M. (2024). Exploring the Role of CCNF Variants in Italian ALS Patients. Genes, 15(12), 1566. https://doi.org/10.3390/genes15121566