
A Novel GBF1 Variant in a Charcot-Marie-Tooth Type 2: Insights from Familial Analysis

 , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
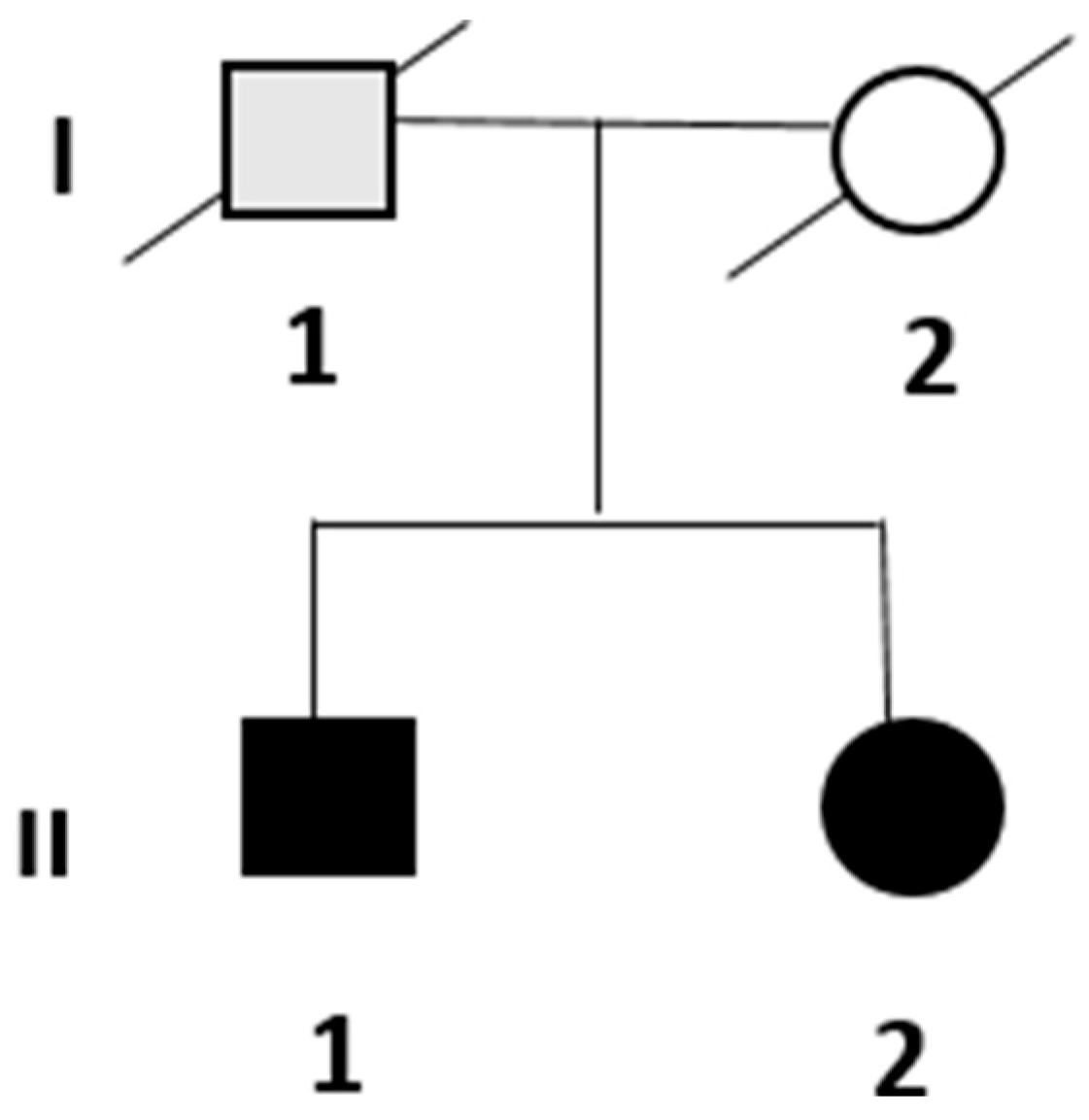
3. Results
3.1. Clinical and Instrumental Assessment
3.1.1. Patient II.1
3.1.2. Patient II.2
3.2. Genetic Analysis
3.3. Diagnosis
4. Discussion
4.1. Clinical Presentation and Genetic Insights
4.2. Limitations and Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harding, A.E.; Thomas, P.K. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain 1980, 103, 259–280. [Google Scholar] [CrossRef] [PubMed]
- Pisciotta, C.; Bertini, A.; Tramacere, I.; Manganelli, F.; Fabrizi, G.M.; Schenone, A.; Tozza, S.; Cavallaro, T.; Taioli, F.; Ferrarini, M.; et al. Clinical spectrum and frequency of Charcot-Marie-Tooth disease in Italy: Data from the National CMT Registry. Eur. J. Neurol. 2023, 30, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Pareyson, D.; Saveri, P.; Pisciotta, C. New developments in Charcot-Marie-Tooth neuropathy and related diseases. Curr. Opin. Neurol. 2017, 30, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Laurá, M.; Pipis, M.; Rossor, A.M.; Reilly, M.M. Charcot-Marie-Tooth disease and related disorders: An evolving landscape. Curr. Opin. Neurol. 2019, 32, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Lupski, J.R. Charcot-Marie-Tooth polyneuropathy: Duplication, gene dosage, and genetic heterogeneity. Pediatr. Res. 1999, 45, 159–165. [Google Scholar] [CrossRef]
- Higuchi, Y.; Takashima, H. Clinical genetics of Charcot-Marie-Tooth disease. J. Hum. Genet. 2023, 68, 199–214. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Barreto, L.C.; Oliveira, F.S.; Nunes, P.S.; de França Costa, I.M.; Garcez, C.A.; Goes, G.M.; Neves, E.L.; de Souza Siqueira Quintans, J.; de Souza Araújo, A.A. Epidemiologic Study of Charcot-Marie-Tooth Disease: A Systematic Review. Neuroepidemiology 2016, 46, 157–165. [Google Scholar] [CrossRef]
- Murphy, S.M.; Herrmann, D.N.; McDermott, M.P.; Scherer, S.S.; Shy, M.E.; Reilly, M.M.; Pareyson, D. Reliability of the CMT neuropathy score (second version) in Charcot-Marie-Tooth disease. J. Peripher. Nerv. Syst. 2011, 16, 191–198. [Google Scholar] [CrossRef]
- Bis-Brewer, D.M.; Gan-Or, Z.; Sleiman, P.; Inherited Neuropathy Consortium; Hakonarson, H.; Fazal, S.; Courel, S.; Cintra, V.; Tao, F.; Estiar, M.A.; et al. Assessing non-Mendelian inheritance in inherited axonopathies. Anesthesia Analg. 2020, 22, 2114–2119. [Google Scholar] [CrossRef]
- Mathis, S.; Goizet, C.; Tazir, M.; Magdelaine, C.; Lia, A.S.; Magy, L.; Vallat, J.M. Charcot-Marie-Tooth diseases: An update and some new proposals for the classification. J. Med. Genet. 2015, 52, 681–690. [Google Scholar] [CrossRef]
- Magy, L.; Mathis, S.; Le Masson, G.; Goizet, C.; Tazir, M.; Vallat, J.M. Updating the classification of inherited neuropathies: Results of an international survey. Neurology 2018, 90, e870–e876. [Google Scholar] [CrossRef]
- Mendoza-Ferreira, N.; Karakaya, M.; Cengiz, N.; Beijer, D.; Brigatti, K.W.; Gonzaga-Jauregui, C.; Fuhrmann, N.; Hölker, I.; Thelen, M.P.; Zetzsche, S.; et al. De Novo and Inherited Variants in GBF1 are Associated with Axonal Neuropathy Caused by Golgi Fragmentation. Am. J. Hum. Genet. 2020, 107, 763–777. [Google Scholar] [CrossRef]
- GeneCards. “GBF1 Gene: Expression.”. GeneCards. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=GBF1#expression (accessed on 22 November 2024).
- García-Mata, R.; Szul, T.; Alvarez, C.; Sztul, E. ADP-ribosylation factor/COPI-dependent events at the endoplasmic reticulum-Golgi interface are regulated by the guanine nucleotide exchange factor GBF1. Mol. Biol. Cell. 2003, 14, 2250–2261. [Google Scholar] [CrossRef]
- Ackema, K.B.; Hench, J.; Böckler, S.; Wang, S.C.; Sauder, U.; Mergentaler, H.; Westermann, B.; Bard, F.; Frank, S.; Spang, A. The small GTPase Arf1 modulates mitochondrial morphology and function. EMBO J. 2014, 33, 2659–2675. [Google Scholar] [CrossRef]
- Magliozzi, R.; Carrero, Z.I.; Low, T.Y.; Yuniati, L.; Valdes-Quezada, C.; Kruiswijk, F.; van Wijk, K.; Heck, A.J.R.; Jackson, C.L.; Guardavaccaro, D. Inheritance of the Golgi Apparatus and Cytokinesis Are Controlled by Degradation of GBF1. Cell Rep. 2018, 23, 3381–3391.e4. [Google Scholar] [CrossRef] [PubMed]
- Geroldi, A.; Mammi, A.; Gaudio, A.; Patrone, S.; La Barbera, A.; Origone, P.; Ponti, C.; Sanguineri, F.; Massucco, S.; Marinelli, L.; et al. Next-generation sequencing in Charcot-Marie-Tooth: A proposal for improvement of ACMG guidelines for variant evaluation. J. Med. Genet. 2024, 61, 847–852. [Google Scholar] [CrossRef]
- Fowler, D.M.; Rehm, H.L. Will variants of uncertain significance still exist in 2030? Am. J. Hum. Genet. 2024, 111, 5–10. [Google Scholar] [CrossRef]
- Zhao, A.; Li, Y.; Niu, M.; Li, G.; Luo, N.; Zhou, L.; Kang, W.; Liu, J. SNPs in SNCA, MCCC1, DLG2, GBF1 and MBNL2 are associated with Parkinson’s disease in southern Chinese population. J. Cell Mol. Med. 2020, 24, 8744–8752. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Knott, A.B.; Bossy-Wetzel, E. Impairing the mitochondrial fission and fusion balance: A new mechanism of neurodegeneration. Ann. NY Acad. Sci. 2008, 1147, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Aerts, M.B.; Weterman, M.A.; Quadri, M.; Schelhaas, H.J.; Bloem, B.R.; Esselink, R.A.; Baas, F.; Bonifati, V.; van de Warrenburg, B.P. A LRSAM1 mutation links Charcot-Marie-Tooth type 2 to Parkinson’s disease. Ann. Clin. Transl. Neurol. 2015, 3, 146–149. [Google Scholar] [CrossRef]
- Stavrou, M.; Kleopa, K.A. Gene therapies for CMT neuropathies: From the bench to the clinic. Curr. Opin. Neurol. 2024, 37, 445–454. [Google Scholar] [CrossRef]

{kind=link}
| Motor | Onset (ms) | Amplitude (mV) | Velocity (m/s) | |||
| II.1 | II.2 | II.1 | II.2 | II.1 | II.2 | |
| Tibial Nerve Right (at knee) | 18.2 | 15.4 | 1.4 | 0.7 | 36.3 | 33.2 |
| Tibial Nerve Left (at knee) | 16.1 | 16.5 | 0.9 | 0.3 | 38.3 | 43.2 |
| Peroneal Nerve Right (below fibula) | 15.45 | 13.9 | 2.2 | 3.1 | 31.9 | 39.1 |
| Peroneal Nerve Left (below fibula) | 16.6 | 13.7 | 3.1 | 2.1 | 31.5 | 39.7 |
| Median Right (at elbow) | - | 8.2 | - | 4.8 | - | 50.0 |
| Sensory | Onset (ms) | Amplitude (mV) | Velocity (m/s) | |||
| II.1 | II.2 | II.1 | II.2 | II.1 | II.2 | |
| Sural Nerve Left (leg) | NR | 3.2 | NR | 10.0 | NR | 46.9 |
| Superficial Peroneal Nerve Right (lateral leg) | NR | 2.5 | NR | 3.5 | NR | 32.0 |
| CMT Neuropathy Score (CMTNS)— Version 2 * | Patient II.1 | Patient II.2 |
|---|---|---|
| Sensory symptoms 0 = None 1 = Symptoms below or at ankle bones 3 = Up to the proximal half of the calf, including knee 4 = Above knee (above the top of the patella) | 3 | 2 |
| Motor symptoms (legs) 0 = None 1 = Trips, catches toes, slaps feet, Shoe inserts 2 = Ankle support or stabilization (AFOs) Foot surgery 3 = Walking aids (cane, walker) 4 = Wheelchair | 2 | 1 |
| Motor symptoms (arms) 0 = None 1 = Mild difficulty with buttons 2 = Severe difficulty or unable to do buttons 3 = Unable to cut most foods 4 = Proximal weakness (affect movements involving the elbow and above) | 0 | 0 |
| Pinprick sensibility 0 = Normal 1 = Decreased below or at ankle bones 2 = Decreased up to the distal half of the calf 3 = Decreased up to the proximal half of the calf, including knee 4 = Decreased above knee (above the top of the patella) | 0 | 1 |
| Vibration 0 = Normal 1 = Reduced at great toe 2 = Reduced at ankle 3 = Reduced at knee (tibial tuberosity) 4 = Absent at knee | 2 | 2 |
| Total Score | 7 | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciampana, V.; Corrado, L.; Magistrelli, L.; Contaldi, E.; Comi, C.; D’Alfonso, S.; Vecchio, D. A Novel GBF1 Variant in a Charcot-Marie-Tooth Type 2: Insights from Familial Analysis. Genes 2024, 15, 1556. https://doi.org/10.3390/genes15121556
Ciampana V, Corrado L, Magistrelli L, Contaldi E, Comi C, D’Alfonso S, Vecchio D. A Novel GBF1 Variant in a Charcot-Marie-Tooth Type 2: Insights from Familial Analysis. Genes. 2024; 15(12):1556. https://doi.org/10.3390/genes15121556
Chicago/Turabian StyleCiampana, Valentina, Lucia Corrado, Luca Magistrelli, Elena Contaldi, Cristoforo Comi, Sandra D’Alfonso, and Domizia Vecchio. 2024. "A Novel GBF1 Variant in a Charcot-Marie-Tooth Type 2: Insights from Familial Analysis" Genes 15, no. 12: 1556. https://doi.org/10.3390/genes15121556
APA StyleCiampana, V., Corrado, L., Magistrelli, L., Contaldi, E., Comi, C., D’Alfonso, S., & Vecchio, D. (2024). A Novel GBF1 Variant in a Charcot-Marie-Tooth Type 2: Insights from Familial Analysis. Genes, 15(12), 1556. https://doi.org/10.3390/genes15121556