

De Novo Genome Assembly and Phylogenetic Analysis of Cirsium nipponicum

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Material and Methods
2.1. Plant Materials, DNA, and RNA Extraction
2.2. Short-Read Illumina Sequencing
2.3. Long-Read Nanopore Sequencing
2.4. Genome Assembly
2.5. Genome Annotation
2.6. Phylogenetic Analyses
2.7. Gene Family Expansion and Contraction Analyses
2.8. Identification of Gene Duplications
3. Results and Discussion
3.1. Genome Sequencing and Assembly
3.2. Repeat Sequence and Gene Prediction
3.3. Comparative Genomic and Phylogenetic Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BUSCO | Benchmarking universal single-copy orthologs |
| BLAST | Basic local alignment search tool |
| Mb | Megabase pairs |
| IsoSeq | Isoform sequencing |
| ONT | Oxford Nanopore Technologies |
| TEs | Transposable elements |
| GO | Gene Ontology |
| Ks | Synonymous substitution rates |
| WGS | Whole genome sequencing |
| LTR | Long terminal repeat |
| Mya | Million years ago |
References
- Kim, B.; Lee, Y.; Koh, B.; Jhang, S.Y.; Lee, C.H.; Kim, S.; Chi, W.-J.; Cho, S.; Kim, H.; Yu, J. Distinctive origin and evolution of endemic thistle of Korean volcanic island: Structural organization and phylogenetic relationships with complete chloroplast genome. PLoS ONE 2023, 18, e0277471. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Lee, K.-R. Phytochemical constituents of Cirsium nipponicum (MAX.) Makino. Korean J. Pharmacogn. 2005, 36, 145–150. [Google Scholar]
- Yin, J.; Heo, S.-I.; Wang, M.-H. Antioxidant and antidiabetic activities of extracts from Cirsium japonicum roots. Nutr. Res. Pract. 2008, 2, 247. [Google Scholar] [CrossRef]
- Lv, Y.; Gao, S.; Xu, S.; Du, G.; Zhou, J.; Chen, J. Spatial organization of silybin biosynthesis in milk thistle [Silybum marianum (L.) Gaertn]. Plant J. 2017, 92, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Dallio, M.; Loguercio, C. Silymarin/Silybin and Chronic Liver Disease: A Marriage of Many Years. Molecules 2017, 22, 191. [Google Scholar] [CrossRef]
- Shaker, E.; Mahmoud, H.; Mnaa, S. Silymarin, the antioxidant component and Silybum marianum extracts prevent liver damage. Food Chem. Toxicol. 2010, 48, 803–806. [Google Scholar] [CrossRef]
- Kim, K.D.; Shim, J.; Hwang, J.-H.; Kim, D.; El Baidouri, M.; Park, S.; Song, J.; Yu, Y.; Lee, K.; Ahn, B.-O. Chromosome-level genome assembly of milk thistle (Silybum marianum (L.) Gaertn.). Sci. Data 2024, 11, 342. [Google Scholar] [CrossRef]
- Vargas, P.; Zardoya, R. Evolution on islands. In The Tree of Life: Evolution and Classification of Living Organisms; Sinauer Associates: Sunderland, MA, USA, 2014; pp. 577–594. [Google Scholar]
- Oh, S.-H.; Chen, L.; Kim, S.-H.; Kim, Y.-D.; Shin, H. Phylogenetic relationship of Physocarpus insularis (Rosaceae) endemic on Ulleung Island: Implications for conservation biology. J. Plant Biol. 2010, 53, 94–105. [Google Scholar] [CrossRef]
- Stuessy, T.F.; Jakubowsky, G.; Gómez, R.S.; Pfosser, M.; Schlüter, P.M.; Fer, T.; Sun, B.Y.; Kato, H. Anagenetic evolution in island plants. J. Biogeogr. 2006, 33, 1259–1265. [Google Scholar] [CrossRef]
- Song, M.-J.; Kim, H. Taxonomic study on Cirsium Miller (Asteraceae) in Korea based on external morphology. Korean J. Plant Taxon. 2007, 37, 17–40. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef] [PubMed]
- Holley, G.; Beyter, D.; Ingimundardottir, H.; Møller, P.L.; Kristmundsdottir, S.; Eggertsson, H.P.; Halldorsson, B.V. Ratatosk: Hybrid error correction of long reads enables accurate variant calling and assembly. Genome Biol. 2021, 22, 28. [Google Scholar] [CrossRef]
- Hu, J.; Wang, Z.; Sun, Z.; Hu, B.; Ayoola, A.O.; Liang, F.; Li, J.; Sandoval, J.R.; Cooper, D.N.; Ye, K. NextDenovo: An efficient error correction and accurate assembly tool for noisy long reads. Genome Biol. 2024, 25, 107. [Google Scholar] [CrossRef]
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. NextPolish: A fast and efficient genome polishing tool for long-read assembly. Bioinformatics 2020, 36, 2253–2255. [Google Scholar] [CrossRef]
- Guan, D.; McCarthy, S.A.; Wood, J.; Howe, K.; Wang, Y.; Durbin, R. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 2020, 36, 2896–2898. [Google Scholar] [CrossRef]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Rodriguez, M.; Makałowski, W. Software evaluation for de novo detection of transposons. Mobile DNA 2022, 13, 14. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, L.; Brůna, T.; Hoff, K.J.; Ebel, M.; Lomsadze, A.; Borodovsky, M.; Stanke, M. BRAKER3: Fully automated genome annotation using RNA-seq and protein evidence with GeneMark-ETP, AUGUSTUS, and TSEBRA. Genome Res. 2024. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef]
- Stanke, M.; Schöffmann, O.; Morgenstern, B.; Waack, S. Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources. BMC Bioinform. 2006, 7, 62. [Google Scholar] [CrossRef]
- Brůna, T.; Lomsadze, A.; Borodovsky, M. GeneMark-ETP significantly improves the accuracy of automatic annotation of large eukaryotic genomes. Genome Res. 2024. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Tang, S.; Lomsadze, A.; Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 2015, 43, e78. [Google Scholar] [CrossRef]
- Hart, A.J.; Ginzburg, S.; Xu, M.; Fisher, C.R.; Rahmatpour, N.; Mitton, J.B.; Paul, R.; Wegrzyn, J.L. EnTAP: Bringing faster and smarter functional annotation to non-model eukaryotic transcriptomes. Mol. Ecol. Resour. 2020, 20, 591–604. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2020, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A Program for Improved Detection of Transfer RNA Genes in Genomic Sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Loman, T. A Novel Method for Predicting Ribosomal RNA Genes in Prokaryotic Genomes. Master’s Thesis, Lund University, Lund, Sweden, 2017. [Google Scholar]
- Griffiths-Jones, S.; Moxon, S.; Marshall, M.; Khanna, A.; Eddy, S.R.; Bateman, A. Rfam: Annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005, 33, D121–D124. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Rannala, B.; Yang, Z. Inferring speciation times under an episodic molecular clock. Syst. Biol. 2007, 56, 453–466. [Google Scholar]
- Fukushima, K.; Pollock, D.D. Detecting macroevolutionary genotype–phenotype associations using error-corrected rates of protein convergence. Nat. Ecol. Evol. 2023, 7, 155–170. [Google Scholar] [CrossRef]
- Puttick, M.N. MCMCtreeR: Functions to prepare MCMCtree analyses and visualize posterior ages on trees. Bioinformatics 2019, 35, 5321–5322. [Google Scholar] [CrossRef] [PubMed]
- Mendes, F.K.; Vanderpool, D.; Fulton, B.; Hahn, M.W. CAFE 5 models variation in evolutionary rates among gene families. Bioinformatics 2020, 36, 5516–5518. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenführer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-h.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef]
- Bai, C.; Alverson, W.S.; Follansbee, A.; Waller, D.M. New reports of nuclear DNA content for 407 vascular plant taxa from the United States. Ann. Bot. 2012, 110, 1623–1629. [Google Scholar] [CrossRef]
- Garcia, S.; Hidalgo, O.; Jakovljević, I.; Siljak-Yakovlev, S.; Vigo, J.; Garnatje, T.; Vallès, J. New data on genome size in 128 Asteraceae species and subspecies, with first assessments for 40 genera, 3 tribes and 2 subfamilies. Plant Biosyst.-Int. J. Deal. All Asp. Plant Biol. 2013, 147, 1219–1227. [Google Scholar] [CrossRef]
- Bureš, P.; Wang, Y.-F.; Horová, L.; Suda, J. Genome size variation in Central European species of Cirsium (Compositae) and their natural hybrids. Ann. Bot. 2004, 94, 353–363. [Google Scholar] [CrossRef]
- Macas, J.; Novák, P.; Pellicer, J.; Čížková, J.; Koblížková, A.; Neumann, P.; Fukova, I.; Doležel, J.; Kelly, L.J.; Leitch, I.J. In depth characterization of repetitive DNA in 23 plant genomes reveals sources of genome size variation in the legume tribe Fabeae. PLoS ONE 2015, 10, e0143424. [Google Scholar] [CrossRef]
- Piegu, B.; Guyot, R.; Picault, N.; Roulin, A.; Saniyal, A.; Kim, H.; Collura, K.; Brar, D.S.; Jackson, S.; Wing, R.A. Doubling genome size without polyploidization: Dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 2006, 16, 1262–1269. [Google Scholar] [CrossRef]
- Neumann, P.; Koblížková, A.; Navrátilová, A.; Macas, J. Significant expansion of Vicia pannonica genome size mediated by amplification of a single type of giant retroelement. Genetics 2006, 173, 1047–1056. [Google Scholar] [CrossRef]
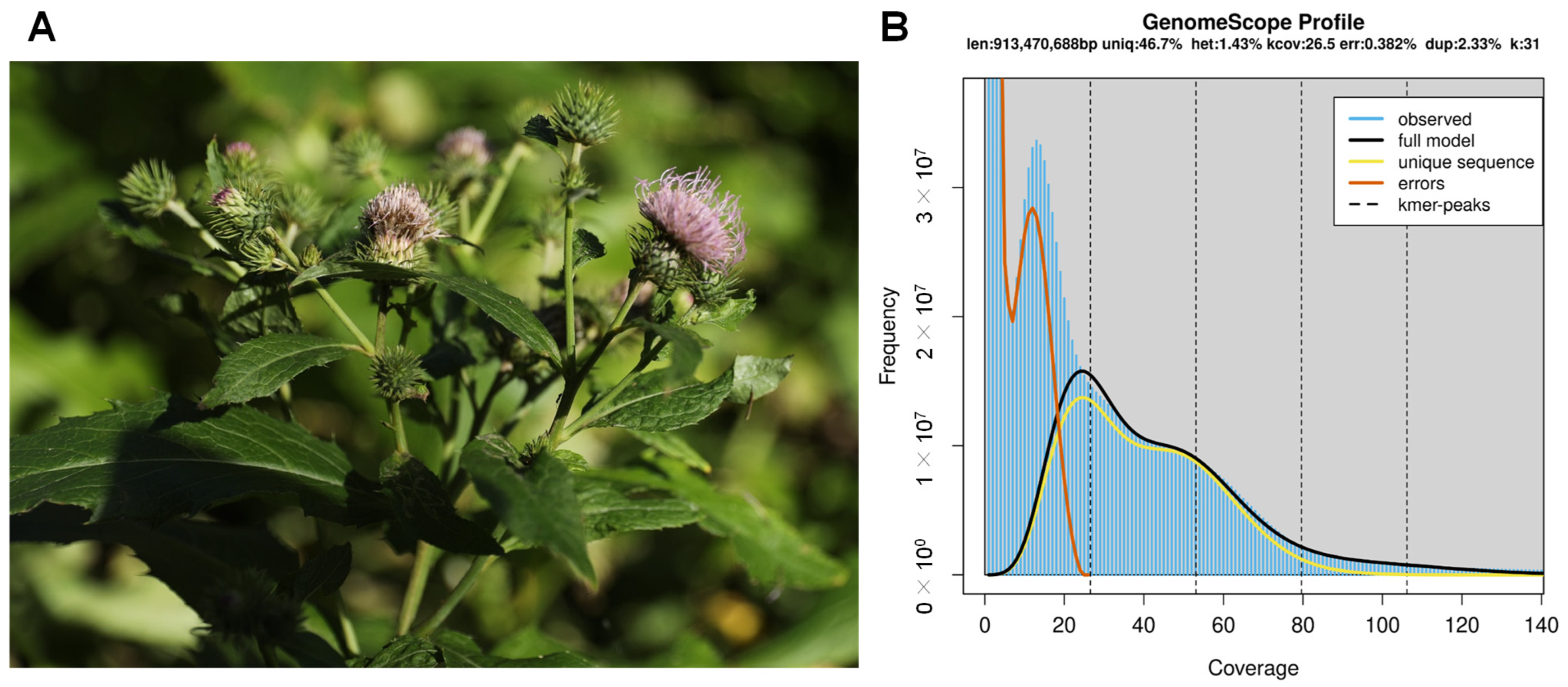
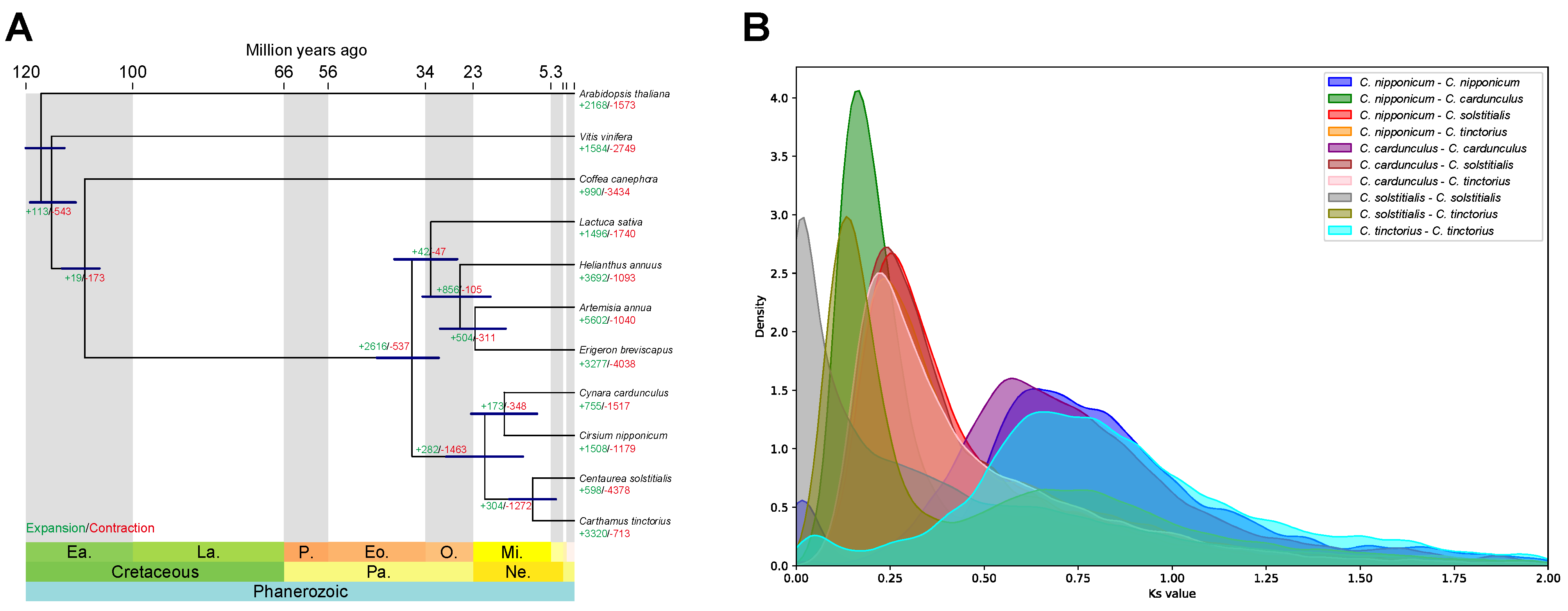


{kind=link}
{kind=link}
{kind=link}
| Genome Assembly | Draft | Purge Haplotigs |
|---|---|---|
| Genome size (Mb) | 1487.8 | 929.4 |
| Number of contigs | 5675 | 2199 |
| N50 (bp) | 421,852 | 700,963 |
| GC contents (%) | 36.01 | 35.76 |
| Number of BUSCOs (%) | ||
|---|---|---|
| Complete | 2212 (95.1) | |
| Complete and single-copy | 1995 (85.8) | |
| Complete and duplicated | 217 (9.3) | |
| Fragmented | 19 (0.8) | |
| Missing | 95 (4.1) |
| Type | Copy | Average Length (bp) | Total Length (bp) |
|---|---|---|---|
| rRNA | 771 | 139.66 | 107,681 |
| tRNA | 1137 | 76.00 | 86,409 |
| miRNA | 159 | 120.81 | 19,208 |
| snRNA | 1907 | 113.67 | 216,769 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, B.Y.; Kim, J.; Park, H.; Kim, J.; Han, S.; Jo, I.-H.; Shim, D. De Novo Genome Assembly and Phylogenetic Analysis of Cirsium nipponicum. Genes 2024, 15, 1269. https://doi.org/10.3390/genes15101269
Choi BY, Kim J, Park H, Kim J, Han S, Jo I-H, Shim D. De Novo Genome Assembly and Phylogenetic Analysis of Cirsium nipponicum. Genes. 2024; 15(10):1269. https://doi.org/10.3390/genes15101269
Chicago/Turabian StyleChoi, Bae Young, Jaewook Kim, Hyeonseon Park, Jincheol Kim, Seahee Han, Ick-Hyun Jo, and Donghwan Shim. 2024. "De Novo Genome Assembly and Phylogenetic Analysis of Cirsium nipponicum" Genes 15, no. 10: 1269. https://doi.org/10.3390/genes15101269
APA StyleChoi, B. Y., Kim, J., Park, H., Kim, J., Han, S., Jo, I.-H., & Shim, D. (2024). De Novo Genome Assembly and Phylogenetic Analysis of Cirsium nipponicum. Genes, 15(10), 1269. https://doi.org/10.3390/genes15101269