1. Introduction
DNA methylation and hydroxymethylation, as well as histone post-translational modifications and non-coding RNAs, constitute the epigenome that regulates chromatin states and gene expression in differentiated cell types of multicellular organisms [
1]. In eukaryotes, DNA methylation occurs at the C5 position of cytosine (5mC), mostly in the context of CG sequences, and plays an important role in gene expression, gene silencing, and genomic imprinting [
2,
3]. Unmethylated CpG islands are present in most human promoters of actively transcribed genes, whereas DNA methylation at the promoters can lead to the transcriptional silencing of the associated genes and at repetitive elements [
4,
5]. DNA methylation is catalyzed by DNA methyltransferases that use S-adenosyl-L-methionine as a methyl group donor [
6]. The active removal of DNA methylation is initiated by the ten-eleven translocation (TET) protein family of dioxygenases, catalyzing the iterative oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) [
7,
8,
9], which is followed by the removal of the oxidized bases 5fC and 5caC catalyzed by Thymine-DNA glycosylase [
10].
During normal development, DNA methylation patterns are established in early embryogenesis and largely maintained afterwards, with small tissue-specific adaptations [
2,
3]. However, in recent years, epigenome editing technologies have been developed, allowing the change in cellular DNA methylation levels at specific target loci. For this, DNA methylation-modifying effector domains are recruited to the desired target region through a DNA-binding domain (DBD). The most flexible and commonly used DBD is the nuclease-deactivated CRISPR-associated protein 9 (dCas9), which can bind to genomic targets via a locus-specific single-guide RNA (sgRNA) sequence [
11]. Instead of fusing a single effector domain to the DBD, it was shown that multiple single-chain variable fragment (scFv)-fused effectors can be recruited to the same genomic locus by using dCas9 fused to a SunTag [
12]. This system was highly efficient and showed a reduced off-target activity in some applications compared to the direct fusion of the DNA methyltransferase DNMT3A effector domain to dCas9 for targeted DNA methylation [
13,
14]. With a modified linker length between the peptide repeats of the original SunTag, this system achieved very strong DNA demethylation efficiencies using TET1CD as the effector domain [
15].
Genomic imprinting is found in about 150 mammalian genes that are frequently involved in growth control and development [
16,
17]. Imprinted genes are solely expressed from either the maternal or the paternal allele. They often occur in clusters, spanning up to several thousand kilobases [
16,
18]. The differential expression is maintained by allele-specific DNA methylation at imprinting control regions (ICRs) which is established in germ cells and maintained after fertilization throughout life. Additionally, the clusters usually contain at least one non-coding RNA typically expressed from the maternal allele [
16,
18]. Errors in the placement or removal of DNA methylation at ICRs can lead to diseases such as cancer, imprinting disorders, and autoimmune, metabolic, or neurological diseases [
19,
20]. A major cause for the imprinting disorder Silver–Russell syndrome (SRS) is DNA hypomethylation at the imprinting control region 1 (ICR1) on chromosome 11p15, controlling the expression of the insulin-like growth factor 2 gene (
IGF2) and the H19 non-coding RNA [
21,
22,
23]. SRS is a sporadic and genetically heterogeneous syndrome characterized by intrauterine growth retardation, underweight, a short statue, as well as body asymmetries and a triangular face shape [
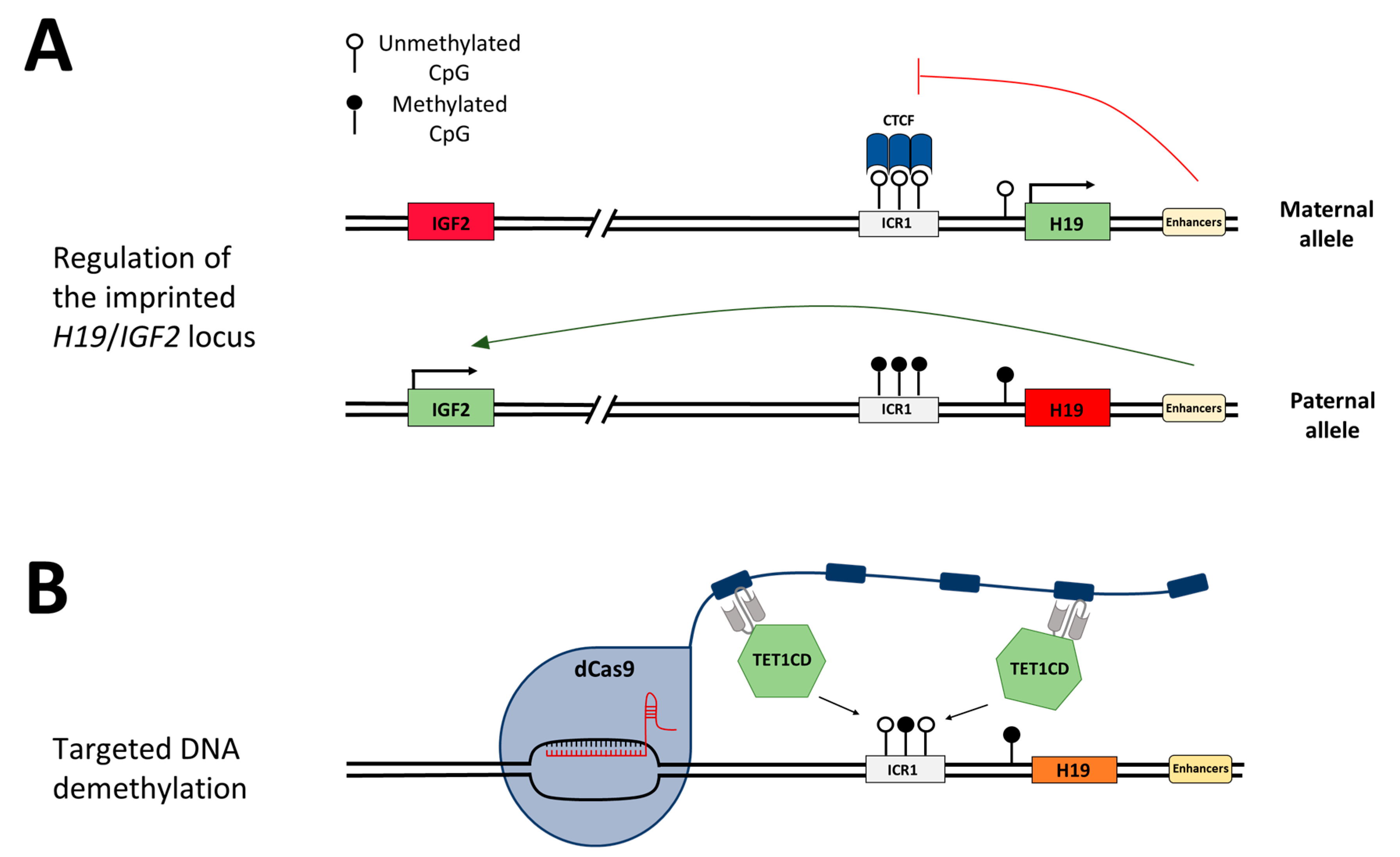
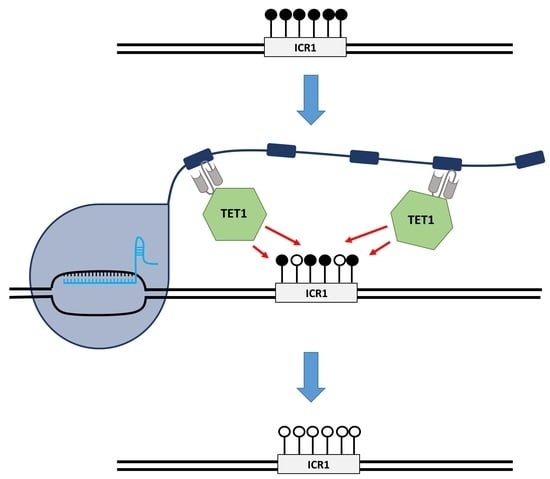
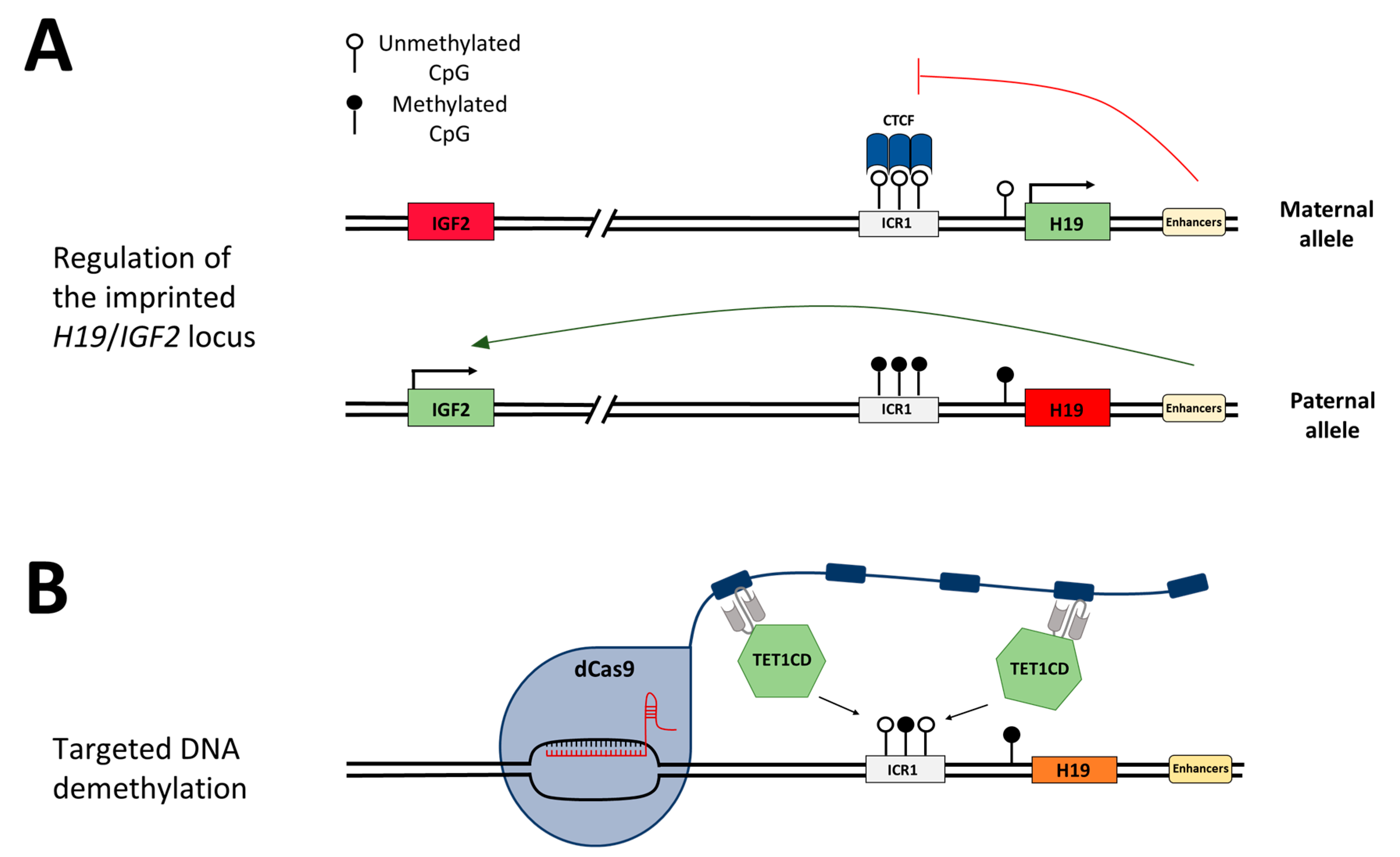
24]. In healthy individuals, the ICR1 is unmethylated on the maternal allele and
H19 is exclusively expressed from this allele (
Figure 1A,B).
IGF2 is silenced through the binding of CTCF (CCCTC-binding factor) to the unmethylated ICR1, which acts as an insulator and hinders downstream enhancers from activating
IGF2 expression. On the paternal allele,
IGF2 is expressed and
H19 is silenced, mediated by DNA methylation at ICR1. The loss of the DNA methylation imprinting mark at the ICR1 in SRS leads to the biallelic repression of
IGF2 and biallelic expression of
H19 [
25]. IGF2 has growth-promoting effects; hence, the loss of its expression explains the clinical features of SRS. The imprinting disorder Beckwith–Wiedemann syndrome (BWS) is, contrary to SRS, associated with increased birth weight and somatic overgrowth. Additionally, BWS leads to a predisposition for embryonal malignancies [
26]. In 5–10% of diagnosed BWS cases, DNA hypermethylation is found at the ICR1, which causes an opposite gene expression to that observed in SRS patients, namely, the downregulation of
H19 and biallelic expression of
IGF2 [
27].
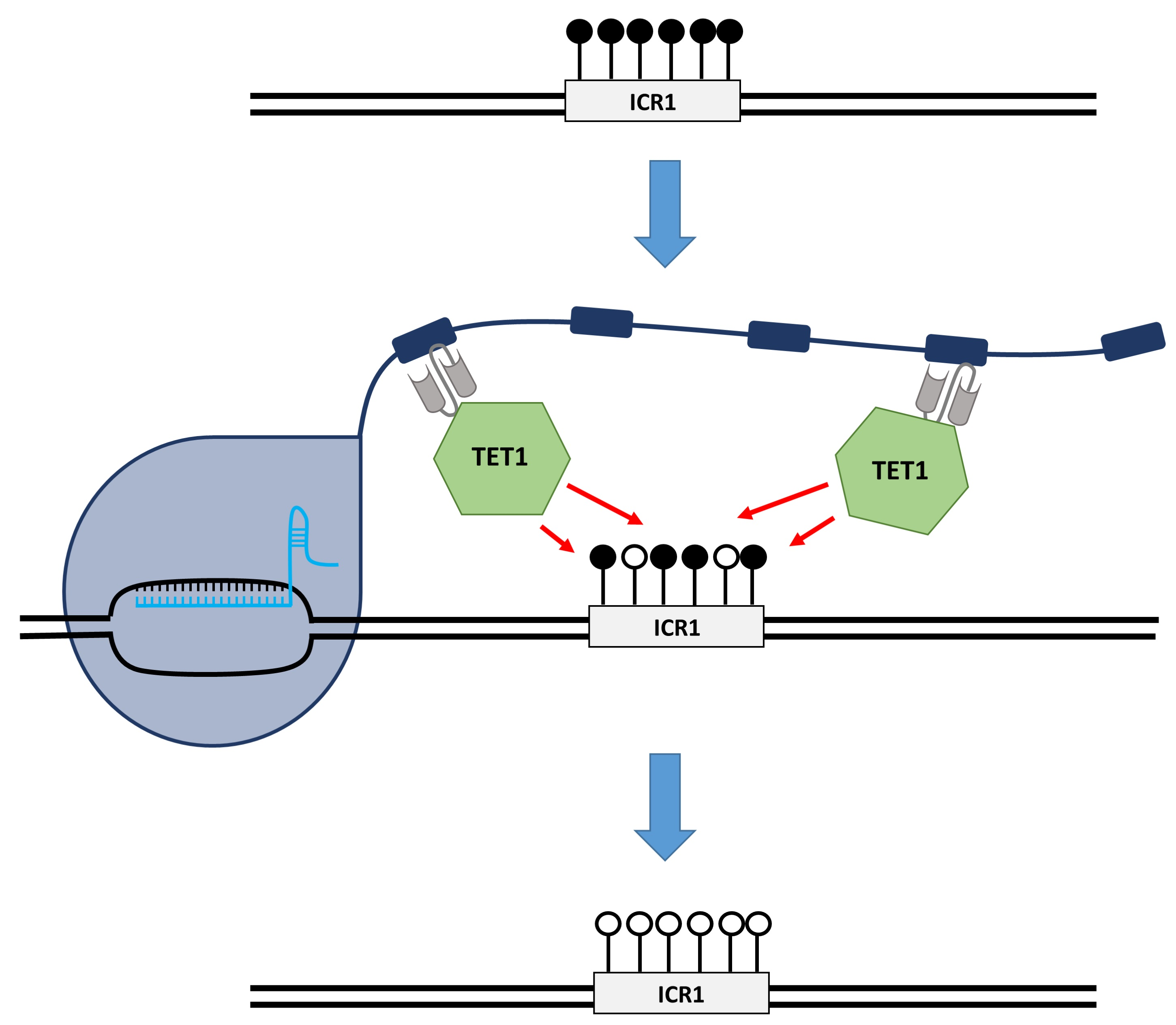
In this study, we aim to demethylate the ICR1 in HEK293 cells to mimic the disease state of SRS and investigate the arising concepts for the treatment of BWS by epigenome editing. For this, we apply an epigenome editing system, in which the target locus is bound by the sgRNA/dCas9 complex fused to the SunTag [
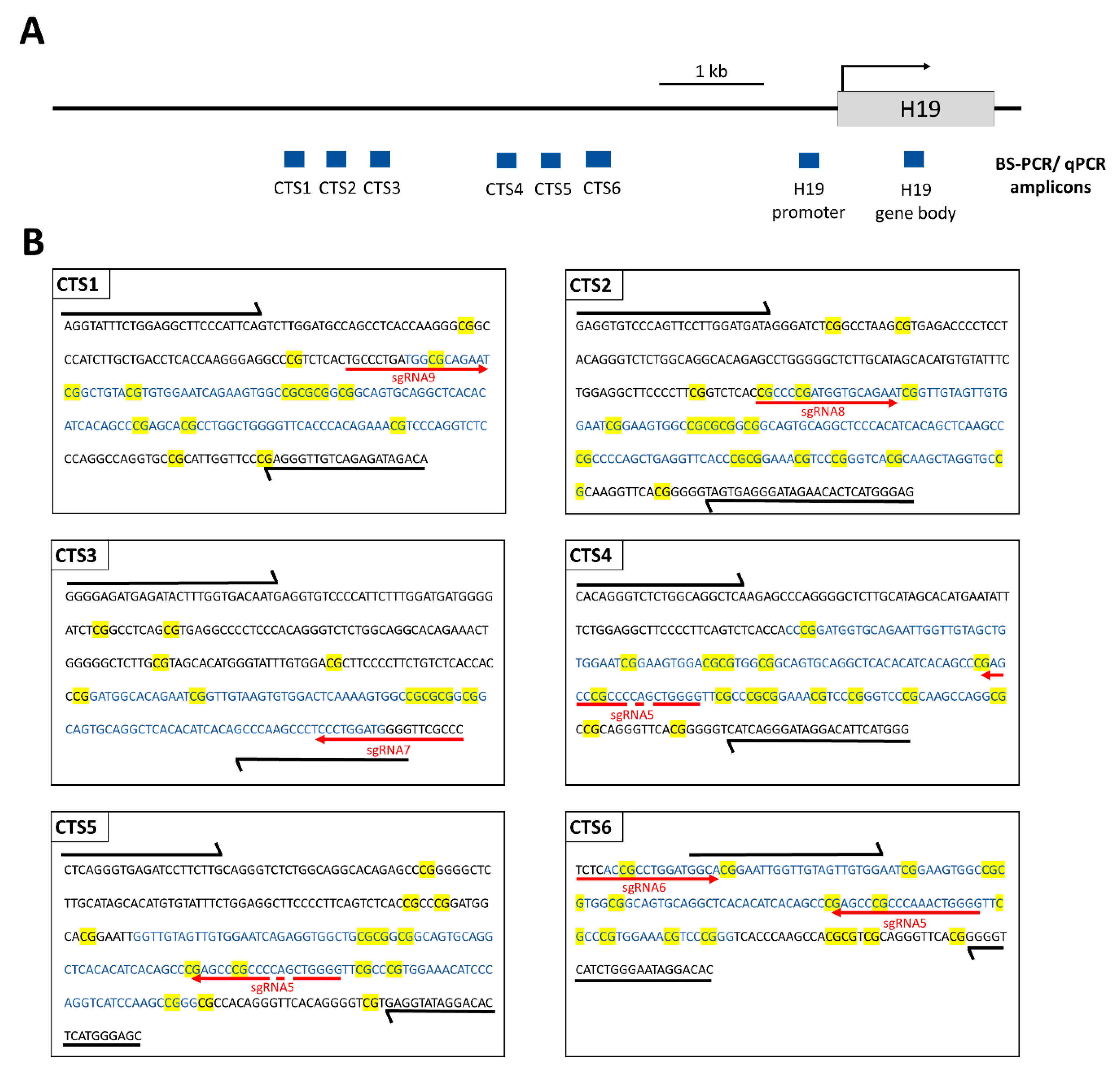
12], which in turn recruits the scFv-fused catalytic domain of TET1 (TET1CD) to finally trigger DNA demethylation. We use a two-plasmid system, one containing dCas9 fused to the SunTag with five repeats of the GCN4 peptide, separated by 22 aa long linkers, and scFv-fused TET1CD. The second plasmid encodes five sgRNAs targeting the ICR1 (
Supplementary Figure S1). The possibility to demethylate the
H19/IGF2 ICR1 was previously shown in two related epigenome editing studies. In 2016, Morita et al. applied a similar epigenome editing system in embryonic stem cells (ESCs) and observed an almost complete demethylation of four CTCF target sites in ICR1 after treatment, resulting in a significant increase in
H19 expression [
15]. In 2020, Horii et al. demonstrated an effective reprogramming of the
H19/
IGF2 ICR1 in a mouse disease model by applying the epigenome editing system to demethylate ICR1, leading to an upregulation of
H19 and downregulation of
IGF2 expression [
25]. In addition, they also applied their system to HEK293 cells. By using six sgRNAs targeting CTCF target -sites (CTSs) and four sgRNAs targeting the
H19 promoter region, they showed significant demethylation at all sgRNA targets five days after transfection [
25]. Again, the expression of
H19 was upregulated and
IGF2 expression was downregulated. Furthermore, at day 9 after transfection, they showed increased CTCF occupancy at two targeted CTCF binding sites, as well as in one targeted
H19 promoter region. In parallel, our lab performed similar studies in HEK293 cells. In this paper, we were able to confirm the previously shown reprogramming of the
H19/
IGF2 imprinting locus. In addition, we document strong demethylation of the ICR1 for up to one month in proliferating HEK293 cells. By applying amplicon-based oxidative bisulfite-sequencing, we demonstrate the appearance of 5hmC at the target regions as a primary editing product generated by the catalytic activity of TET1CD. The CTCF occupancy at all targeted CTSs was permanently increased. We also show an increase in
H19 expression up to day 27 after transfection. Likewise, the downregulation of
IGF2 expression was observed three days after transfection; however, this reduction was not maintained until day 27.
2. Materials and Methods
2.1. Cloning of the EpiEditor Constructs
The individual sgRNA sequences (
Supplementary Table S1) were firstly cloned into the single sgRNA expression vector (Addgene plasmid #210212), and then all five sgRNA sequences together with individual U6 promoters and sgRNA scaffolds were cloned into the multi-sgRNA vector (pMulti-sgRNA-LacZ-DsRed vector, a gift from Yujie Sun, Addgene plasmid #99914), as previously described [
28]. For the dCas9-5×SunTag-scFV-TET1CD vector, the dCas9-5×SunTag part was taken from the Addgene plasmid #82560, into which scFv-IRES-GFP was cloned to create the dCas9-5×SunTag-scFv-empty vector. The TET1CD sequence was amplified from the Addgene plasmid #82561 by PCR and inserted into the dCas9-5×SunTag-scFv-empty vector to create the final dCas9-5×SunTag-scFv-TET1CD vector (
Supplementary Figure S1).
2.2. Cell Culture
Human Embryonic Kidney cells 293 (HEK293) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% Fetal Bovine Serum (FBS) (Sigma-Aldrich, Taufkirchen, Germany), 4 mM L-glutamine (Sigma-Aldrich, Taufkirchen, Germany), and 10 mL/L penicillin/streptomycin (Sigma-Aldrich, Taufkirchen, Germany). The cells were incubated at 37 °C, 95% relative humidity, and 5% CO2. The cell confluency was maintained under 90% by splitting the cells every 2 or 3 days.
For transfection experiments, 1.4 million cells were seeded into 100 mm cell culture dishes. On the following day, 9000 ng of dCas9-5×SunTag-scFv-TET1CD or of the control plasmid with no effector domain (dCas9-5×SunTag-scFv-empty), 500 ng of sgRNA plasmid (
Supplementary Figure S1), and 27 μL of FuGENE HD transfection reagent (Promega, Walldorf, Germany) were mixed in 840 μL serum-free DMEM. After incubating the mixture for 15 min at RT, it was homogenously distributed across the dish. On the following day, the growth medium was replaced with a freshly supplemented DMEM. Three days after transfection, the cells were trypsinized and filtered through a 30 μm pre-separation filter (Miltenyi Biotec, Bergisch Gladbach, Germany). Living, single cells showing both GFP and DsRed fluorescence signals were sorted using the Cell Sorter SF800S (Sony Biotechnology, San Jose, CA, USA). Approximately 0.5–1 × 10
6 sorted HEK293 cells were used for immediate downstream analysis or cell pellets were snap-frozen with liquid nitrogen and stored at −80 °C. The remaining cells were re-seeded into 6-well plates to harvest at later time points. The harvested cells were subjected to centrifugation for 5 min with 300×
g, the pellets were washed once with PBS and the cell pellets were snap-frozen with liquid nitrogen and stored at −80 °C.
2.3. MBD2-Pulldown-qPCR
Basically, MBD2-pulldown was conducted as described in a previous publication from our laboratory with slight modifications [
29]. In detail, genomic DNA (gDNA) was isolated from cell pellets of 0.5–1 million cells using the QIAmp DNA Mini Kit (QIAGEN GmbH, Hilden, Germany), according to the manufacturer’s instructions. The isolated gDNA (approx. 3–10 µg in a 200 µL volume) was sheared using the EpiShear
TM probe sonicator (Active Motif, Waterloo, Belgium) (2 mm tip, 40% amplitude, 20 × 20 s pulse, and 30 s pause between each pulse). Subsequently, the sheared gDNA was purified using the NucleoSpin
® Gel and PCR Clean-up kit (Macherey-Nagel, Düren, Germany) and eluted in 50 μL ddH
2O pre-warmed to 70 °C. Per sample, 1 μg of sonicated gDNA and 222 nM of GST-MBD2 protein were mixed in cold binding buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 0.5% IGEPAL
® CA-630, and 2 mM DTT) in a final volume of 250 μL, followed by overnight rotation at 4 °C. On the next day, 50 μL Glutathione Agarose 4B beads (Macherey-Nagel, Düren, Germany) were washed four times with 200 μL cold binding buffer (1 min, 2000×
g); the supernatant was removed and 50 µL beads were added to each GST-MBD2/gDNA sample. The beads/GST-MBD2/gDNA mixtures were incubated on a rotating wheel at 4 °C for 2 h. After the incubation, the supernatant was removed by centrifugation for 2 min at 2000×
g. The samples were washed three times with 200 μL cold wash buffer (50 mM Tris-HCl pH 8.0, 500 mM NaCl, 1 mM EDTA, 0.5% IGEPAL
® CA-630, and 2 mM DTT) by rotation for 5 min at 4 °C, followed by centrifugation for 2 min at 2000×
g. The supernatant was discarded, 150 μL of elution buffer (10 mM Tris-HCl pH 8.0, 2000 mM NaCl, and 1 mM EDTA) was added, and the samples were rotated at RT for 15 min. The samples were centrifuged for 2 min at 2000×
g and the supernatant was stored. The elution step was repeated with 100 μL elution buffer and the supernatant was pooled with the first elution fraction. Using the ChIP DNA Purification Kit (Active Motif, Waterloo, Belgium), the DNA was purified. Elution was performed with 100 μL of the elution buffer provided in the ChIP DNA Purification Kit (Active Motif, Waterloo, Belgium).
The DNA was quantified relative to the input DNA (sonicated gDNA) by qPCR using the CFX96 Real-Time PCR detection system (Bio-Rad Laboratories, Inc., Feldkirchen, Germany). A 1:5 dilution series of input gDNA was prepared for each sample. For each qPCR reaction, 7.5 μL 2× ORA™ See qPCR Probe Mix (highQu, Kraichtal, Germany), 0.4 μL forward primer (10 µM), 0.4 μL reverse primer (10 µM), and 5.7 μL ddH
2O were used, to which 1 µL of template DNA was added. The qPCR primers are listed in
Supplementary Table S1. A non-template control (NTC) was included for every primer pair and each sample was pipetted in technical triplicates. The qPCR program was as follows: 95 °C for 3 min, 40 cycles of 95 °C for 3 s, 57 °C for 20 s, and 72 °C for 4 s, followed by a 65–95 °C ramp (0.5 °C increase per 5 s) to generate a melting curve. The qPCR signals were normalized to input and subsequently to the
SLC6A3 locus, which is fully methylated in HEK293 cells [
29].
2.4. CTCF-ChIP-qPCR
Crosslinked chromatin immunoprecipitation (ChIP) followed by qPCR was used to quantify the binding of CTCF at the H19/IGF2 ICR. Four million cells were harvested and pelleted by 300× g for 5 min. The cell pellets were washed twice in PBS and crosslinked by incubating in a solution of 1% formaldehyde in PBS for 10 min, under constant rotation. Afterwards, the reaction was quenched by adding glycine to a final concentration of 125 mM and rotating at RT for 10 min. The crosslinked cells were pelleted and washed with ice-cold PBS. The cell pellets were snap-frozen in liquid nitrogen and stored at −80 °C.
CTCF-ChIP was performed using the Magna ChIP™ HiSens kit (Catalog No. 17-10460, Merck KGaA, Darmstadt, Germany), according to the manufacturer’s protocol. Isolated crosslinked chromatin from 4 million cells was sheared using the EpiShearTM probe sonicator (Active Motif, Waterloo, Belgium) with the 2 mm tip (40% amplitude, 12 × 20 s pulse, and 30 s pause between each pulse). For each ChIP reaction, 50 or 100 μL of crosslinked and sonicated chromatin and 3 μL of αCTCF antibody (CTCF rabbit AB, Cell Signaling Technology Europe, Frankfurt am Main, Germany, catalog #2899S, Lot: 2) were used. As a control, a rabbit IgG of the same isotype (Normal Rabbit IgG Control, catalog #AB-105-C, 1 μg/μL, R&D Systems, Inc./Bio-Techne GmbH, Wiesbaden-Nordenstadt, Germany) was included. The precipitated DNA was quantified relative to the input DNA by qPCR as described for MBD2-pulldown-qPCR. Subsequently, the qPCR signals were normalized to the average of two control CTCF target sites.
2.5. RT-qPCR
Frozen pellets of 0.5–1 million transfected and sorted HEK293 cells were thawed on ice and the RNA was isolated using the RNeasy
® Plus Mini Kit (QIAGEN GmbH, Hilden, Germany), according to the manufacturer’s instructions. To synthesize the complementary DNA (cDNA), 500 ng of purified RNA was used with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems/Life Technologies GmbH, Darmstadt, Germany). Instead of 10× RT Random Primers, the Oligo d(T)
18 mRNA Primer (5 A
260 units, NEB) was used. For each sample, a reaction with reverse transcriptase (plus-RT) and a control reaction without reverse transcriptase (non-RT) were carried out. For the subsequent qPCR, each sample was diluted 1:3 with ddH
2O. For each RT-qPCR reaction, 7.5 μL 2× ORA™ See qPCR Probe Mix (highQu, Kraichtal, Germany), 0.4 μL forward primer (10 µM), 0.4 μL reverse primer (10 µM), and 4.7 μL ddH
2O were mixed, followed by the addition of 2 µL cDNA. The RT-qPCR primers are listed in
Supplementary Table S1. All samples (plus-RT, non-RT, and non-template controls) were pipetted in triplicate. Plus-RT cDNA samples were pooled and a 1:5 input dilution series was prepared to determine the PCR efficiency. The qPCR was run as follows: 95 °C for 3 min, 40 cycles of 95 °C for 15 s, followed by 57 °C (
SDHA) or 62 °C (
H19 and
IGF2) for 30 s, 95 °C for 10 s, and lastly a 65–95 °C ramp (0.5 °C steps every 5 s) to generate the melting curve. The transcript levels of
H19 and
IGF2 were analyzed by normalizing the Cq values to the reference gene Succinate Dehydrogenase Complex Flavoprotein Subunit A (
SDHA) using the −ΔΔCq calculation as described by Pfaffl (2001) [
30].
2.6. Bisulfite Sequencing and Oxidative Bisulfite Sequencing
The gDNA of transfected HEK293 cells sorted by FACS was isolated using the QIAmp DNA Mini Kit (QIAGEN GmbH, Hilden, Germany), according to the manufacturer’s instructions. A total of 500 ng gDNA was fragmented enzymatically by overnight digestion using 40 U EcoRV-HF (a non-cutter in the genomic regions desired for amplification) (New England Biolabs GmbH, Frankfurt am Main, Germany) in CutSmart buffer in a total volume of 20 µL. On the next day, bisulfite conversion was conducted using the EZ DNA Methylation-Lightning™ Kit (Zymo Research Europe GmbH, Freiburg, Germany), according to the manufacturer’s protocol. Oxidative bisulfite conversion was performed using the TrueMethyl® oxBS Module (Part No. 0414, Tecan Group Ltd., Männedorf, Switzerland.), according to the manufacturer’s instructions. Bisulfite-converted DNA was eluted in 12 µL ddH2O.
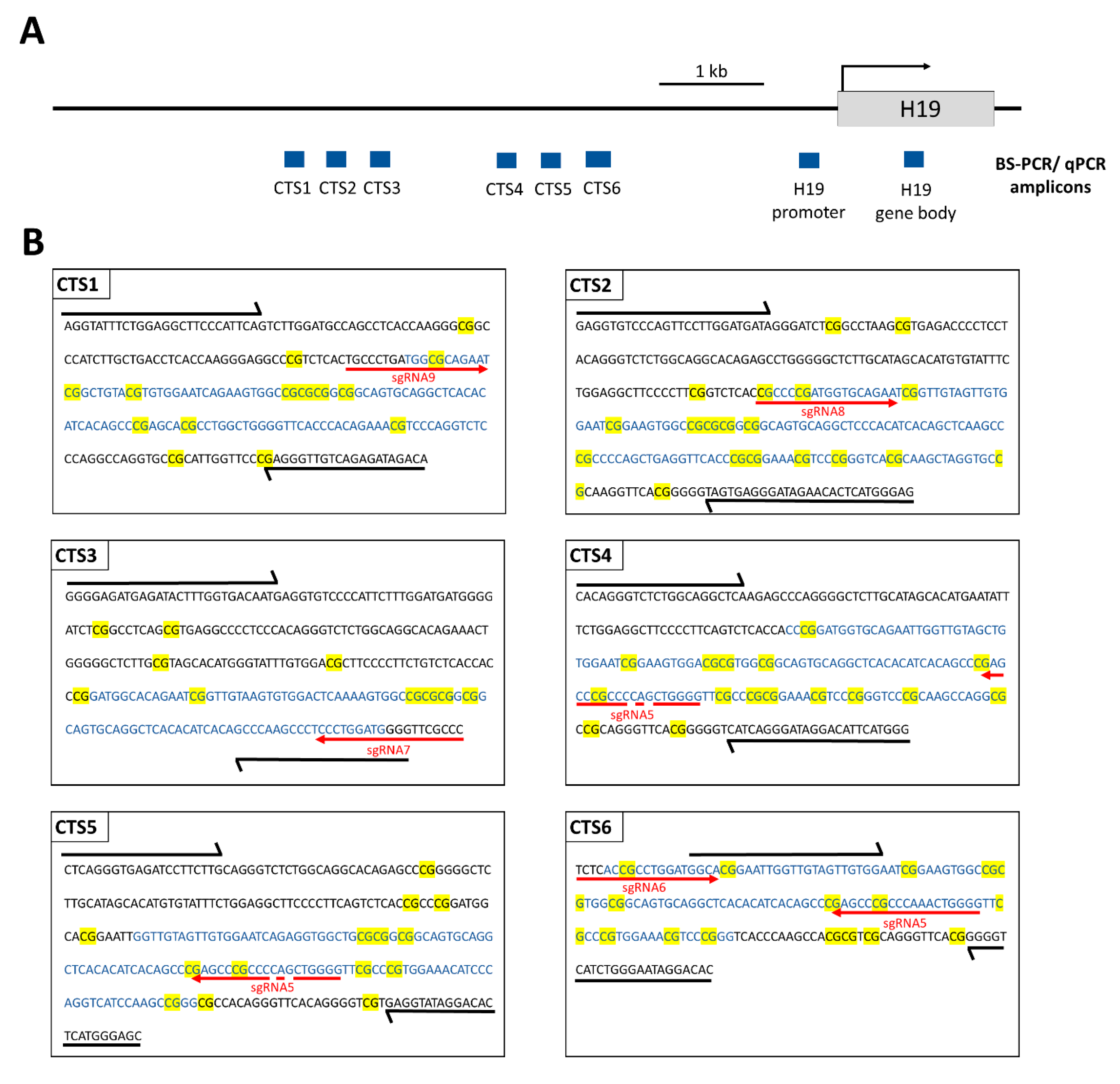
The amplicons of interest were amplified in a first PCR1 with locus-specific primers (
Supplementary Table S1), which also contained barcodes and adapters complementary to PCR2 primers. For each PCR1 reaction, 1 µL of BS-converted DNA template was mixed with 1 µL forward primer (10 µM), 1 µL reverse primer (10 µM), 14.4 µL ddH
2O, 2 µL 10× PCR buffer (QIAGEN GmbH, Hilden, Germany), 0.4 µL dNTPs mix (40 mM), and 0.2 µL of HotStarTaq-Polymerase (5 U/µL, QIAGEN GmbH, Hilden, Germany). The PCR thermocycling program was 95 °C for 15 min, 35 cycles of 94 °C for 30 s, Tm for 30 s, and 72 °C for 1 min, followed by 72 °C for 10 min (Tm values are specified in
Supplementary Table S2). The amplicons were verified by loading 5 µL of the PCR1 reaction on a 1% agarose gel. The PCR1 product was used as a template for PCR2, in which Illumina TruSeq sequencing indices were added to the amplicons. A total of 1 µL PCR1 product (for most genes, the PCR1 product was diluted with ddH
2O before proceeding with PCR2) was mixed with 0.8 µL forward primer (10 µM), 0.8 µL reverse primer (10 µM), 12.8 µL ddH
2O, 4 µL 5× Q5 reaction buffer (New England Biolabs GmbH, Frankfurt am Main, Germany), 0.4 µL dNTPs mix (40 mM), and 0.2 µL of Q5 High-Fidelity DNA Polymerase (New England Biolabs GmbH, Frankfurt am Main, Germany). The PCR thermocycling program was 98 °C for 30 s, 15 cycles of 98 °C for 10 s and 72 °C for 40 s, and lastly 72 °C for 2 min. The PCR2 products were verified by loading 5 µL on a 1% agarose gel. For the final library pooling, the PCR2 amplicons were loaded onto a 0.8% agarose gel run in TAE buffer (1 mM EDTA disodium salt, 40 mM Tris, and 20 mM acetic acid). The bands were excised from the gel and purified using the NucleoSpin
® Gel and PCR Clean-up kit (Macherey-Nagel, Düren, Germany), according to the manufacturer’s protocol. Sample concentrations were measured using the NanoDrop 1000 (Thermo Fisher Scientific/Life Technologies GmbH, Darmstadt, Germany) and equimolar amounts of samples were pooled. Paired-end Illumina sequencing with a 250 bp read length was performed by the Novogene (UK) Company Limited, Cambridge, United Kingdom.
2.7. NGS Data Processing and Data Analysis
NGS data in a FASTQ format were analyzed as described [
28] on the Galaxy platform (
https://usegalaxy.org/) [
31], where all the following tools are available. In brief, Illumina adapter sequences were removed using Trim Galore! (Galaxy Version 0.4.3.1). Afterwards, two paired-end reads were merged using Pear (Galaxy Version 0.9.6.1) and reads with low quality were removed using Filter FASTQ (Galaxy Version 1.1.1). The de-multiplexing of individual samples tagged with combinations of barcodes and Illumina indices was conducted by converting the FASTQ files using FASTQ to Tabular (Galaxy Version 1.1.5), followed by the selection of lines with the tool Select (Galaxy Version 1.0.1) and the re-conversion of the files to a FASTQ format with Tabular to FASTQ (Galaxy Version 1.1.1). For the alignment of reads to a reference sequence, bwameth (Galaxy Version 0.2.0.4) was used and the DNA methylation at each CpG site was analyzed by applying the tool MethylDackel (Galaxy Version 0.3.0.1). The output files were processed using Microsoft Excel (Professional Plus 2019). DNA methylation at non-CpG sites is known to be much lower in human cell lines and it was not investigated in this paper [
32].
4. Discussion
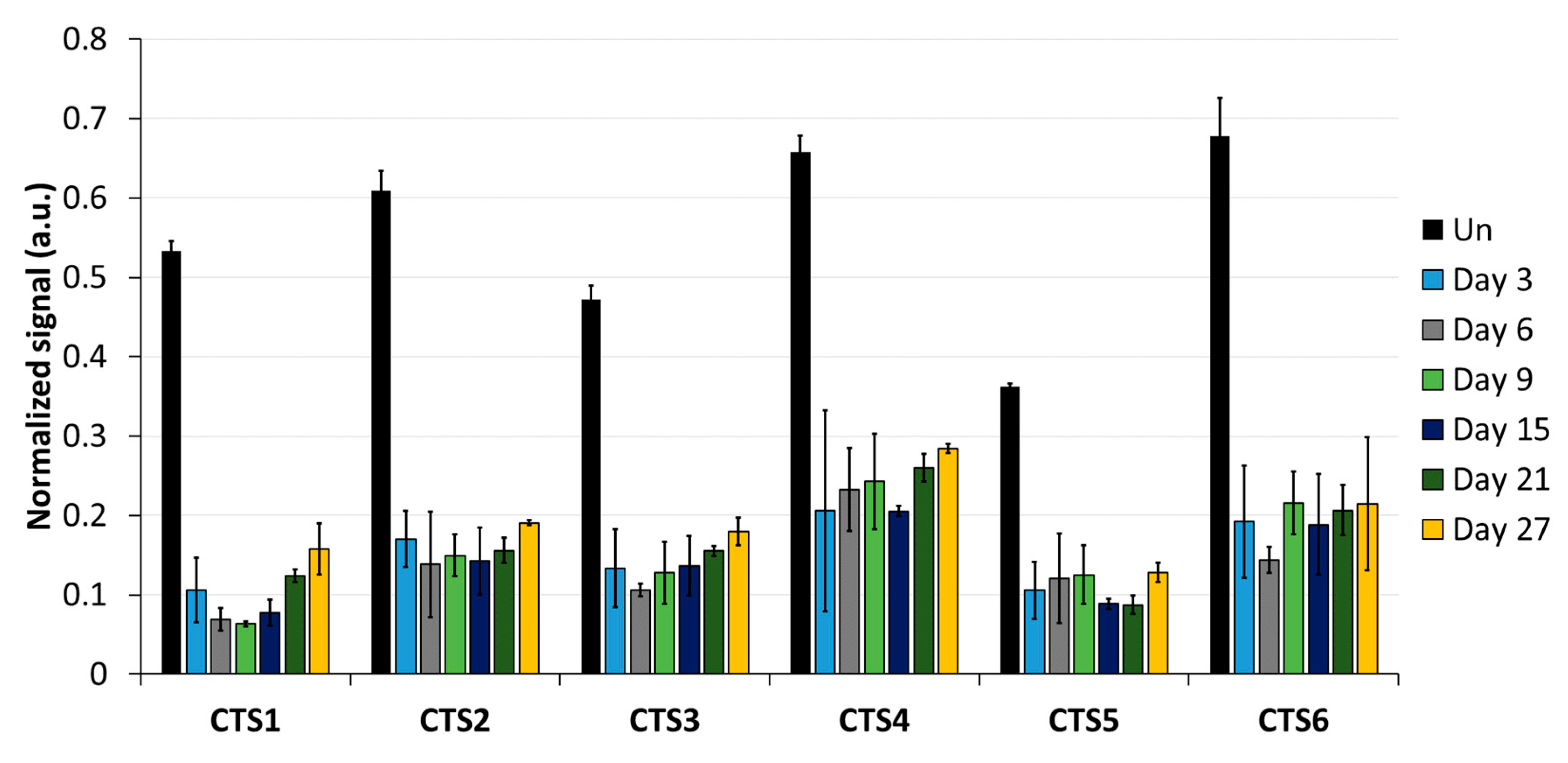
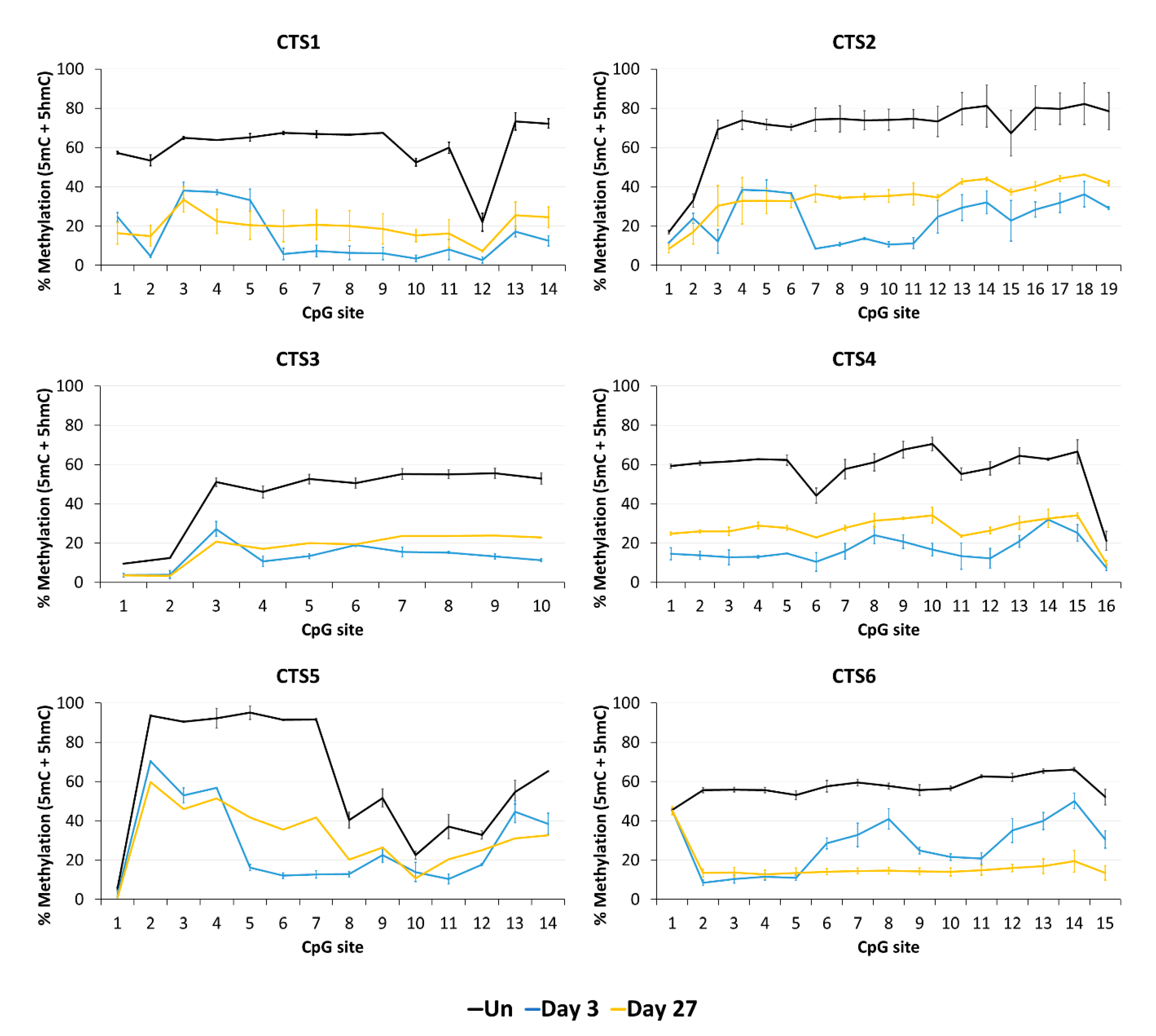
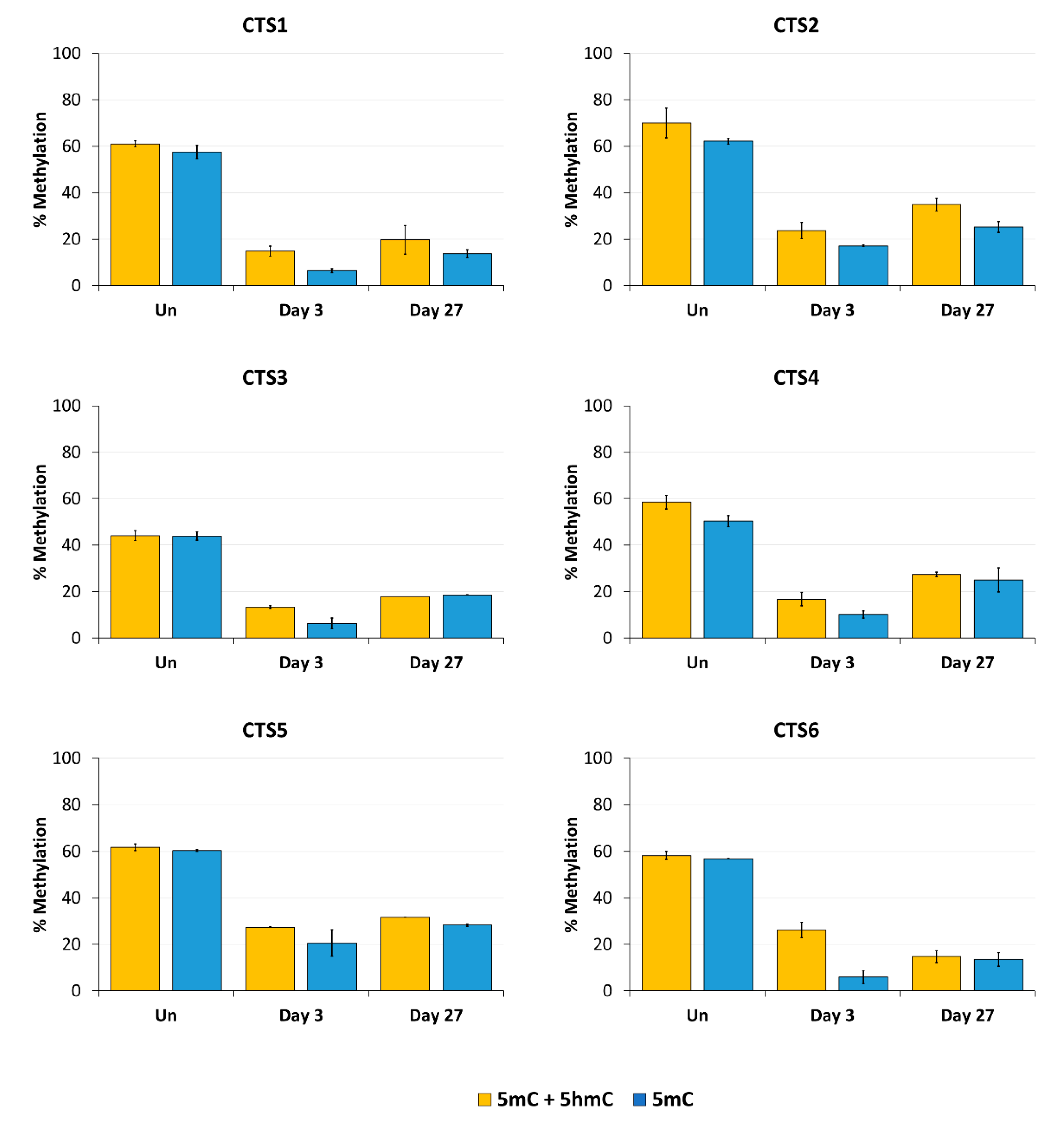
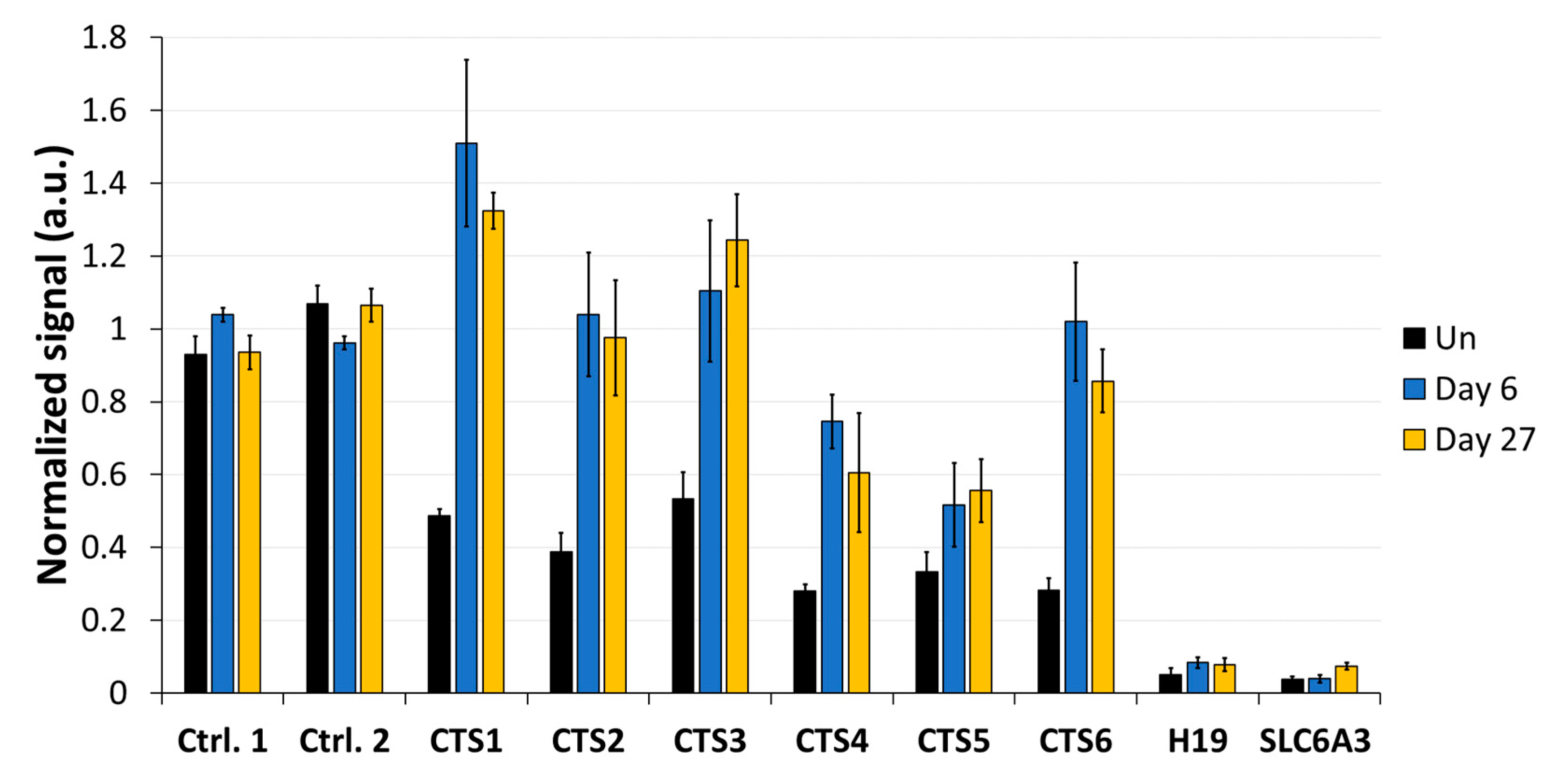
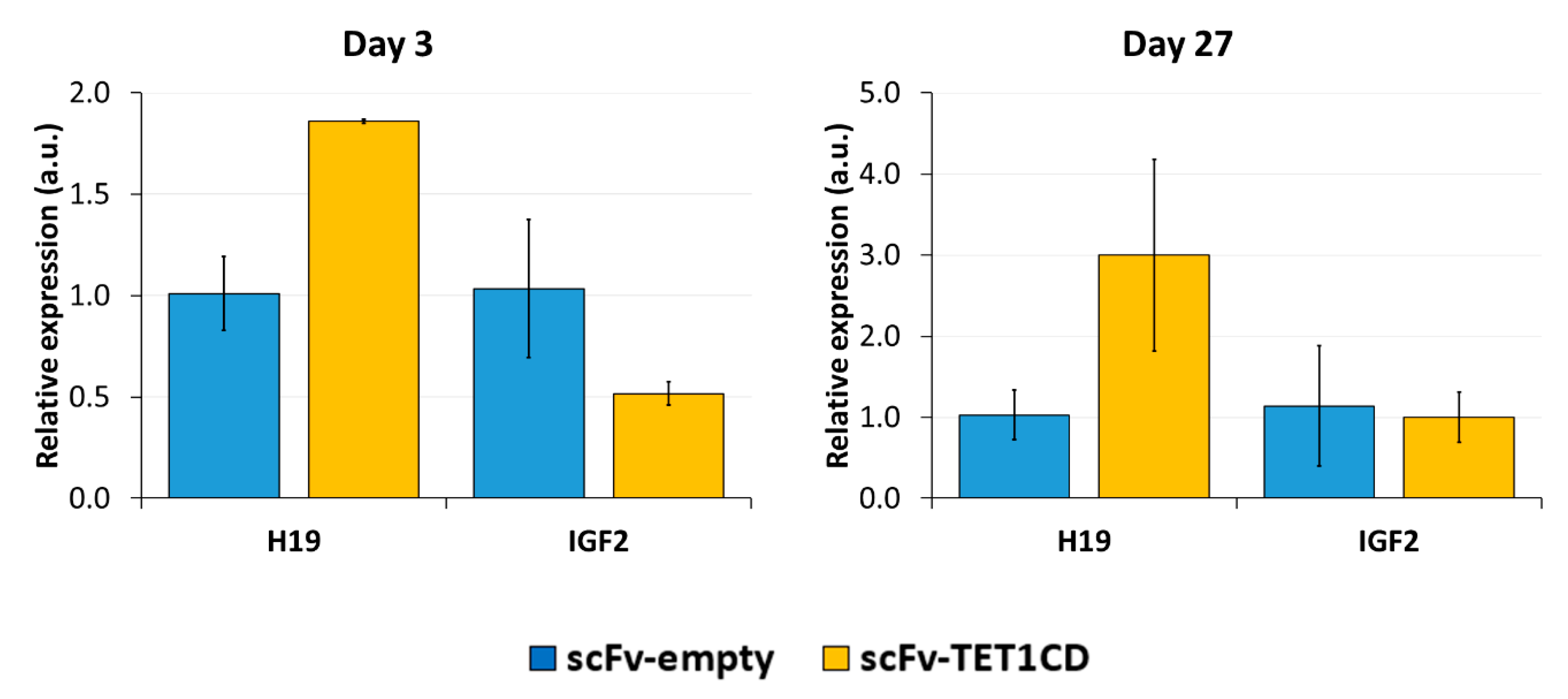
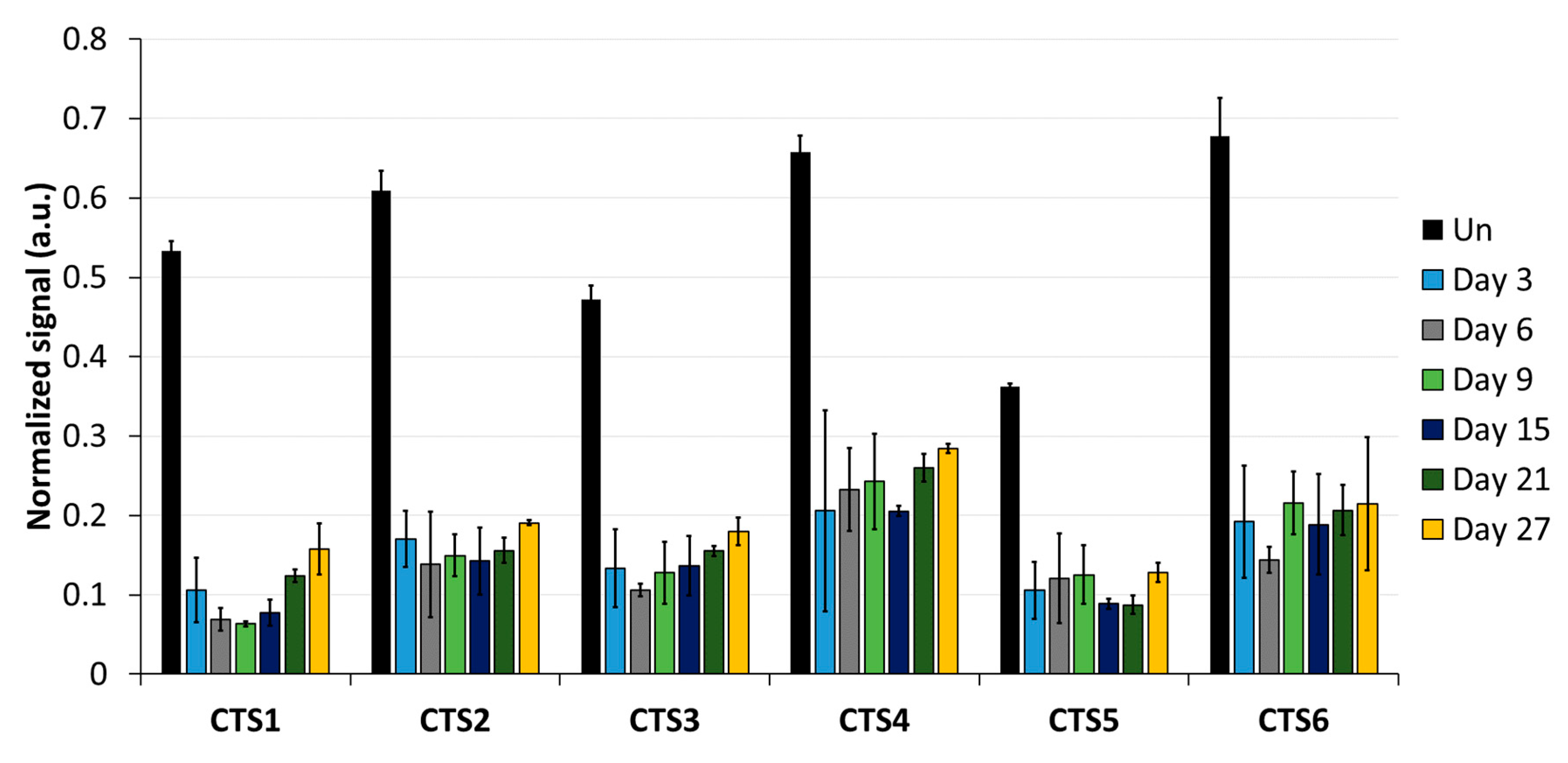
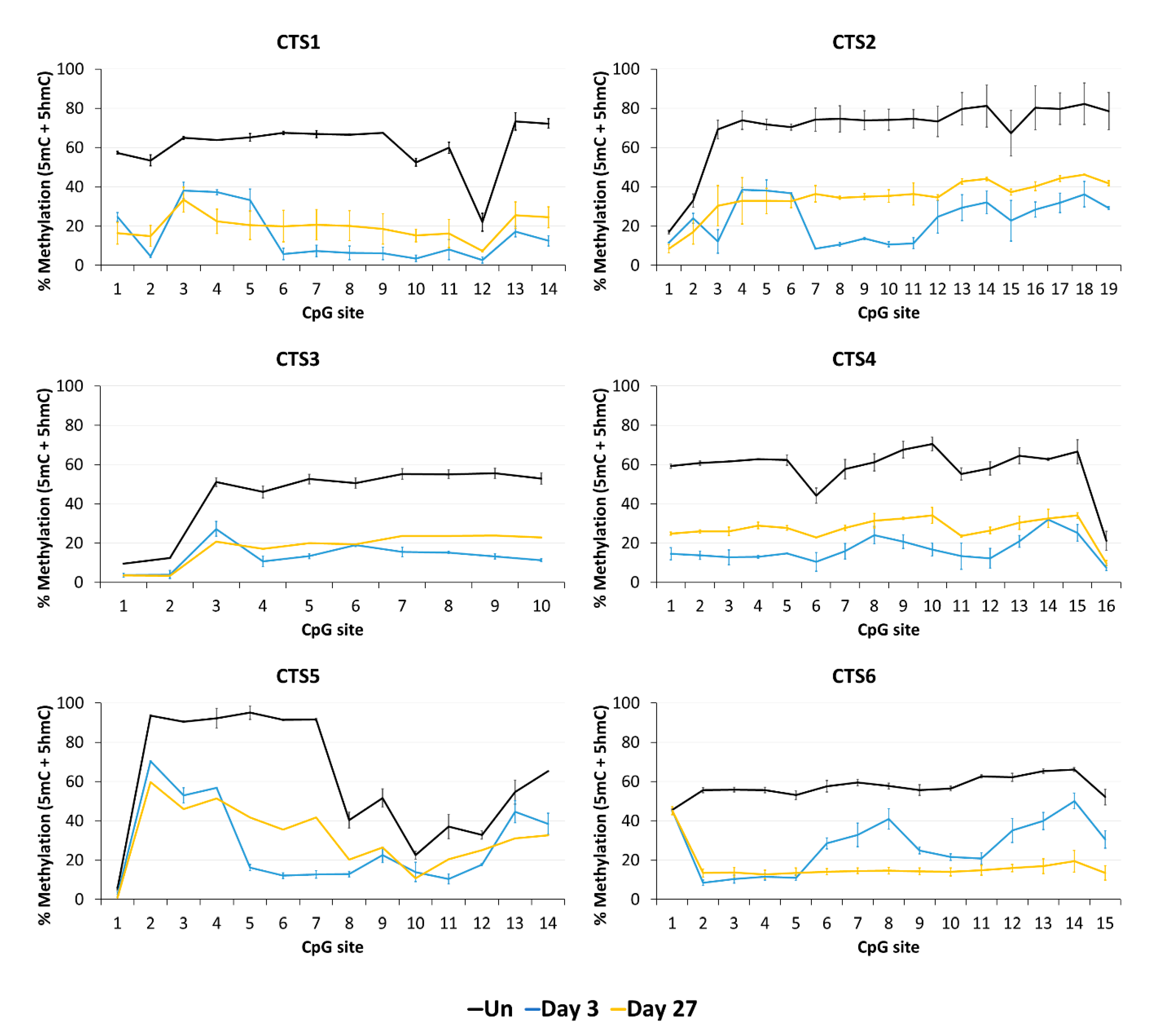
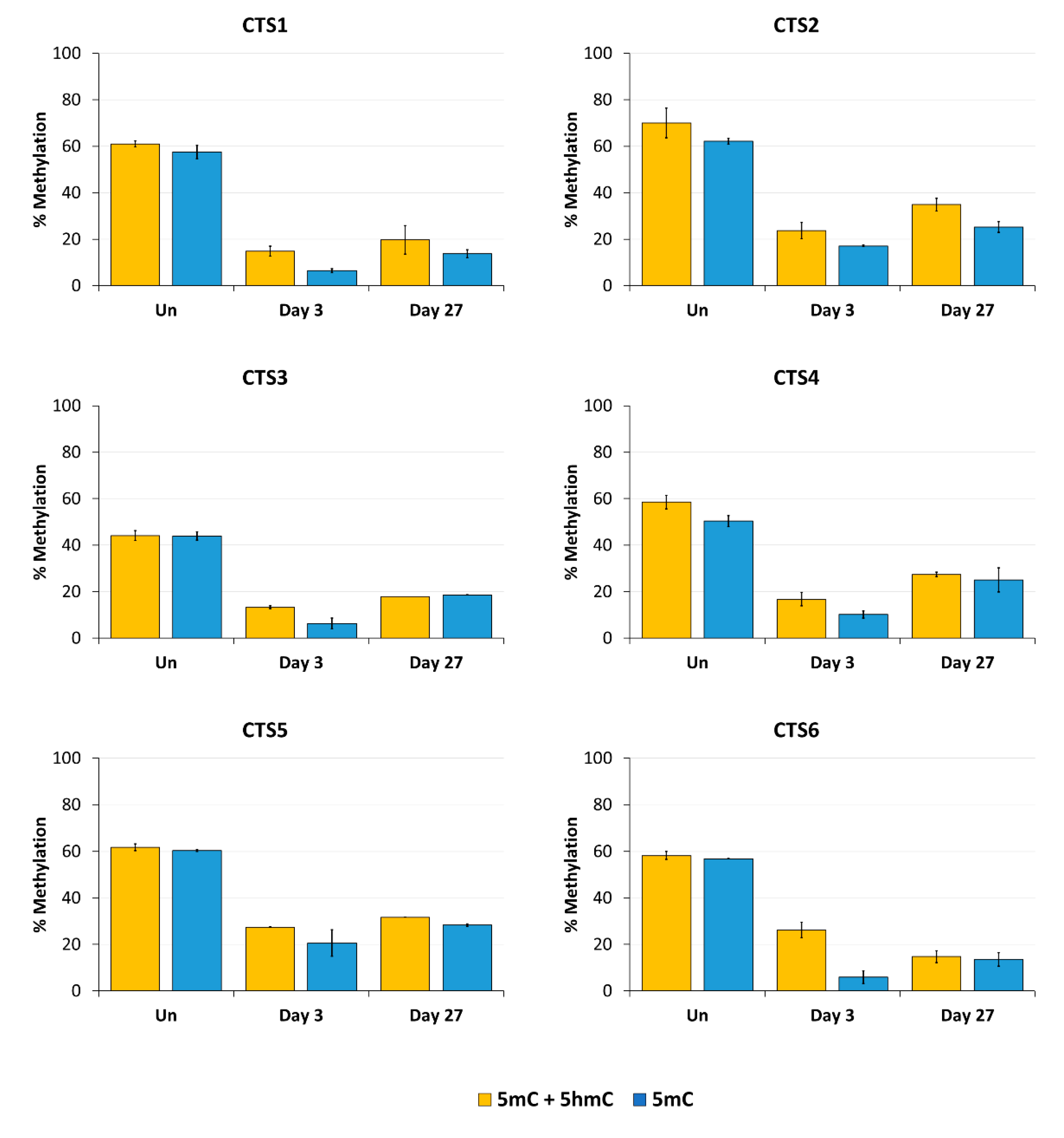
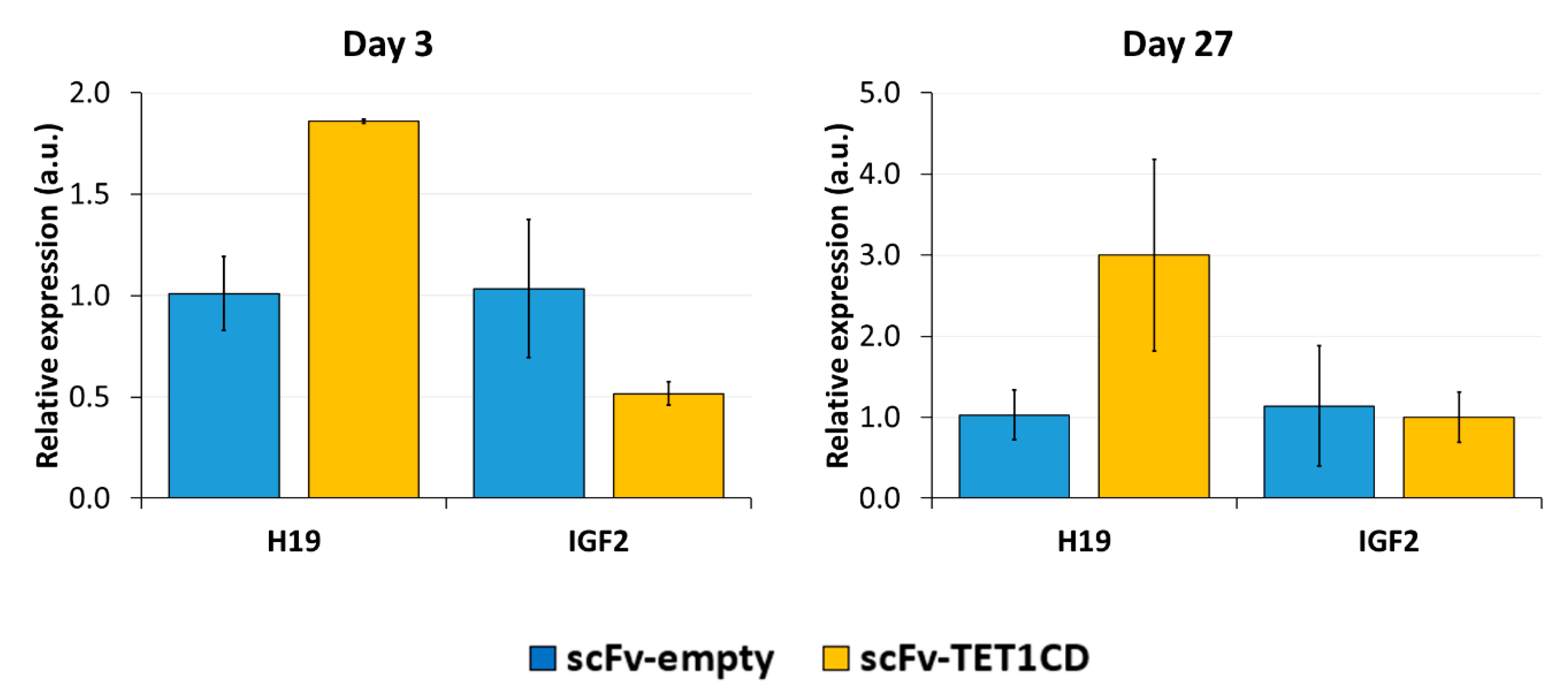
Imprinting is a paradigmatic epigenetic process in which allele-specific DNA methylation at ICRs established in the germ line regulates the allele-specific expression of a set of associated genes in the next generation. Abnormal imprinting can cause rare human diseases, which in principle could be tackled by the targeted rewriting of the DNA methylation at the ICR. Hence, the reprogramming of imprinted loci has interesting clinical applications, but it is also useful for fundamental research. In this study, we conducted a pilot study and successfully demethylated CTCF target sites within the H19/IGF2 ICR1 in human HEK293 cells. We observed the specific generation of 5hmC at the target regions followed by an up to 90% reduction in the 5mC level achieved on day 3 after transfection. Although the expression of the scFv-TET1CD was lost 9 days after transfection, DNA demethylation was stable up to 27 days at the CTS1-6 (the last time point investigated by us), demonstrating the stable epigenome reprogramming of the locus. If one assumes that active editing stopped on day 9 (which is a conservative estimation, as all components must be present in order for the EpiEditor to be active), demethylation was stable for at least 18 days, corresponding to 14–18 cell divisions. Consistent with the robust demethylation, a stable increase in bound CTCF at CTS1-6 was shown and H19 expression was increased until day 27. These findings indicate that not only the DNA methylation was reprogrammed, but the chromatin state of the ICR1 region was altered from a “paternal” (methylated) state in half of the alleles before treatment to a “maternal” (unmethylated) state with CTCF binding in all alleles after treatment. Related to this, it would be interesting to investigate in future if the ICRs change their position in the cell nucleus after epigenome editing, which could be detected by fluorescence in situ hybridization (FISH).
In contrast to the stable expression changes of H19, the expression of IGF2 was reduced only transiently on day 3, but after 27 days, it returned to the expression level observed in the empty-vector control transfection. This effect may represent a secondary response of the cells to reprogramming during continuous culturing, where selective pressure may enrich cells that re-express the IGF2 growth factor and, therefore, propagate faster. In follow-up studies, it would be interesting to study transient and stable gene expression changes at global level after epigenome editing by RNA-seq.
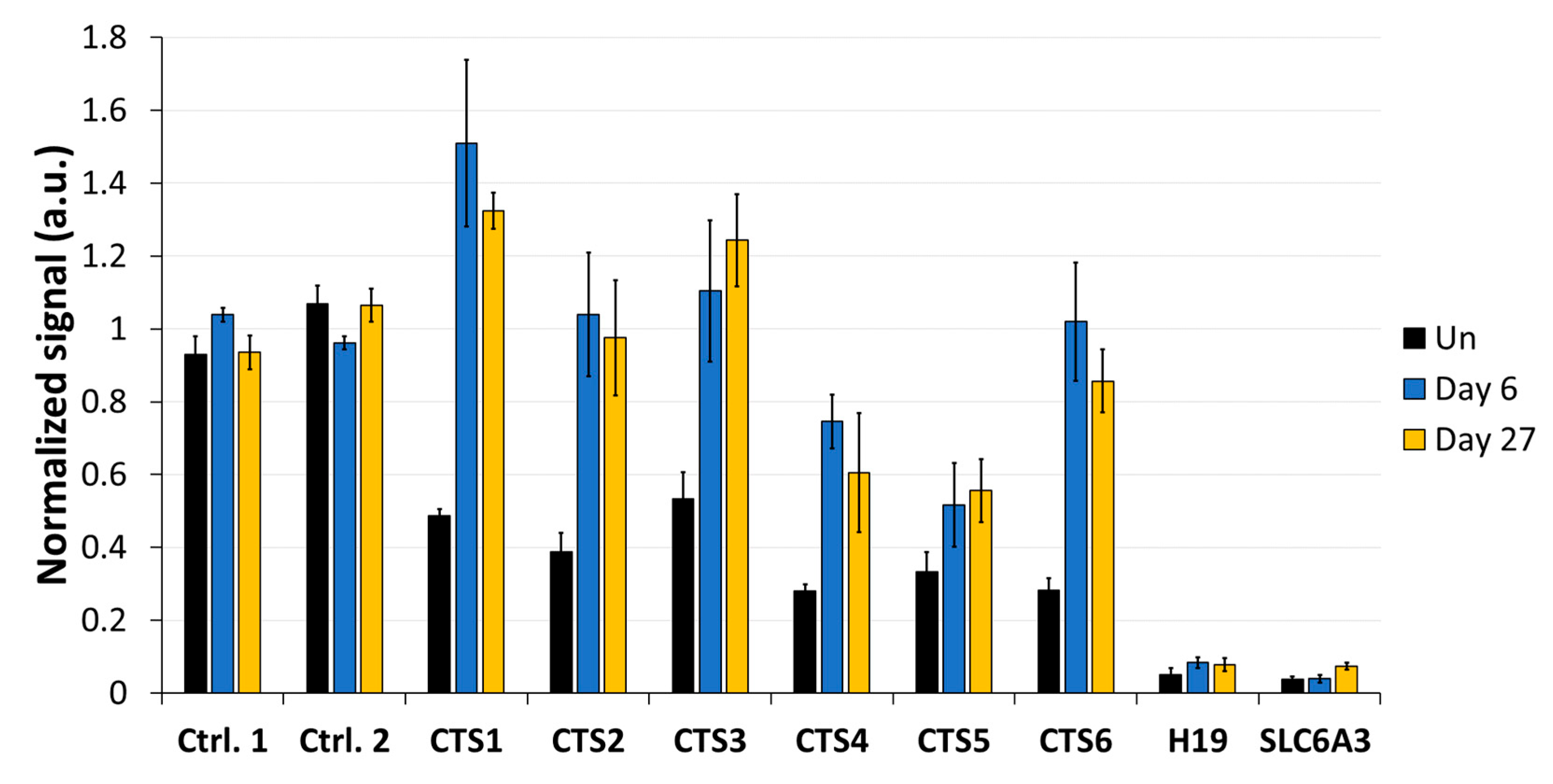
In addition to a robust reprogramming mediated by the epigenome editing tool, it is important to minimize off-target activities, especially if the editors are to be used in clinical applications. We observed low off-target activities at distal sites, which were mostly transient compared to the robust on-target editing. Within the ICR, we observed strong demethylation effects at CTS4 and CTS5, which are targeted by sgRNA5 with two mismatches. Interestingly, we observed a partial DNA demethylation at the
H19 promoter at a distance of more than 1 kb away from the closest sgRNA-binding site, which was more pronounced than the off-target effects observed at the other two tested loci. This finding may be explained by the fact that the ICR1 and
H19 promoter are placed in one topologically associated domain (TAD) in the original ICR1-methylated state when no CTCF is bound [
37]. Therefore, the catalytic domains of TET1 targeted to the ICR1 may also have access to the
H19 promoter due to the flexibility of the targeting device and the chromatin.
Our data are in general agreement with the basic findings of previous studies [
15,
25], but they also provide additional details regarding the reprogramming of the
H19/
IGF2 ICR1 in human HEK293 cells, including the analysis of the CpG site-specific profiles of 5mC and 5hmC and an analysis period of up to 27 days, which provide important new mechanistic insights.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}