
Biological Aging Acceleration Due to Environmental Exposures: An Exciting New Direction in Toxicogenomics Research




Abstract
:1. Introduction
2. Part 1. Biological Clocks as Biomarkers of Aging
2.1. Search Strategy and Selection Criteria
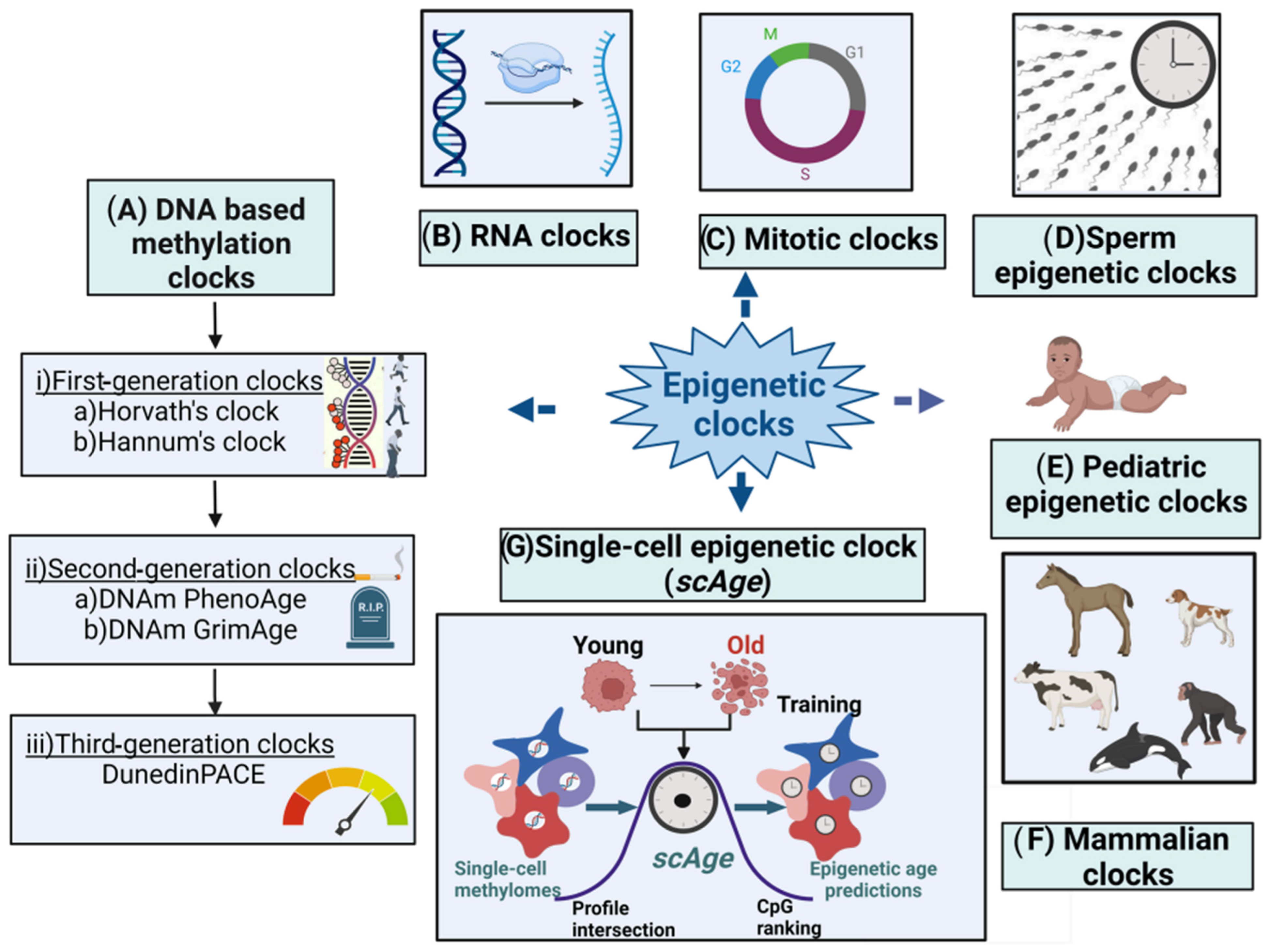
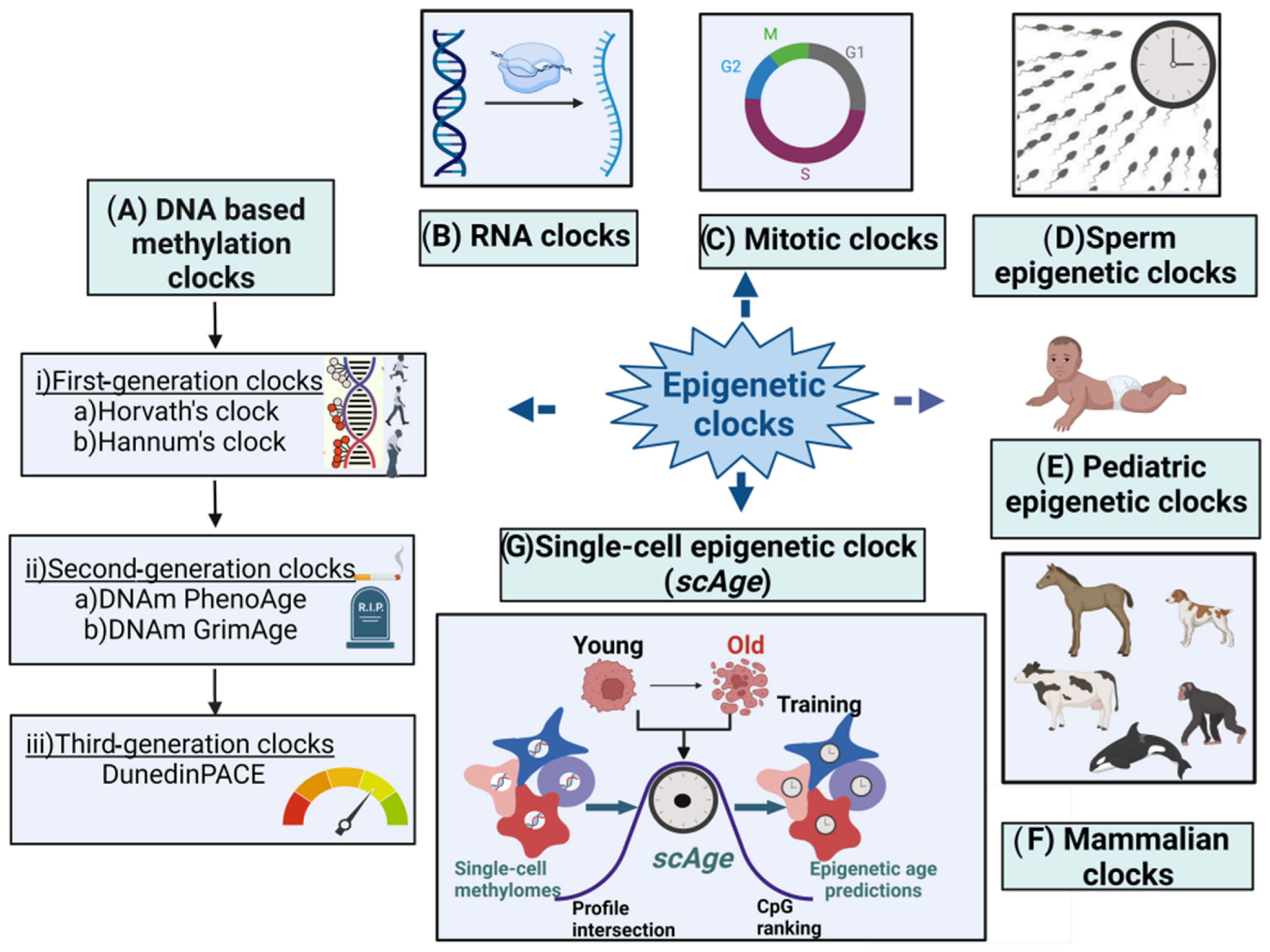
2.2. Epigenetic Clocks
2.3. First-Generation Molecular Epigenetic Clocks
2.3.1. DNA Methylation-Based Molecular Epigenetic Clocks
2.3.2. Horvath’s Clock
2.3.3. Hannum’s Clock
2.4. Second-Generation Epigenetic Clocks
2.4.1. DNAm PhenoAge
2.4.2. DNAm-Based Biomarker of Mortality (DNAm GrimAge)
2.5. Third-Generation Epigenetic Clocks
2.6. Mitotic Clocks
2.7. Sperm Epigenetic Clocks
2.8. Single-Cell Epigenetic Clock Framework (scAge)
2.9. RNA Clocks
2.9.1. RNAAgeCalc: A Multi-Tissue Transcriptional Age Calculator
2.9.2. Multi-Tissue RNA Clock (MultiTIMER)
2.10. Pediatric Epigenetic Clocks
2.11. Nutrition and Epigenetic Clocks
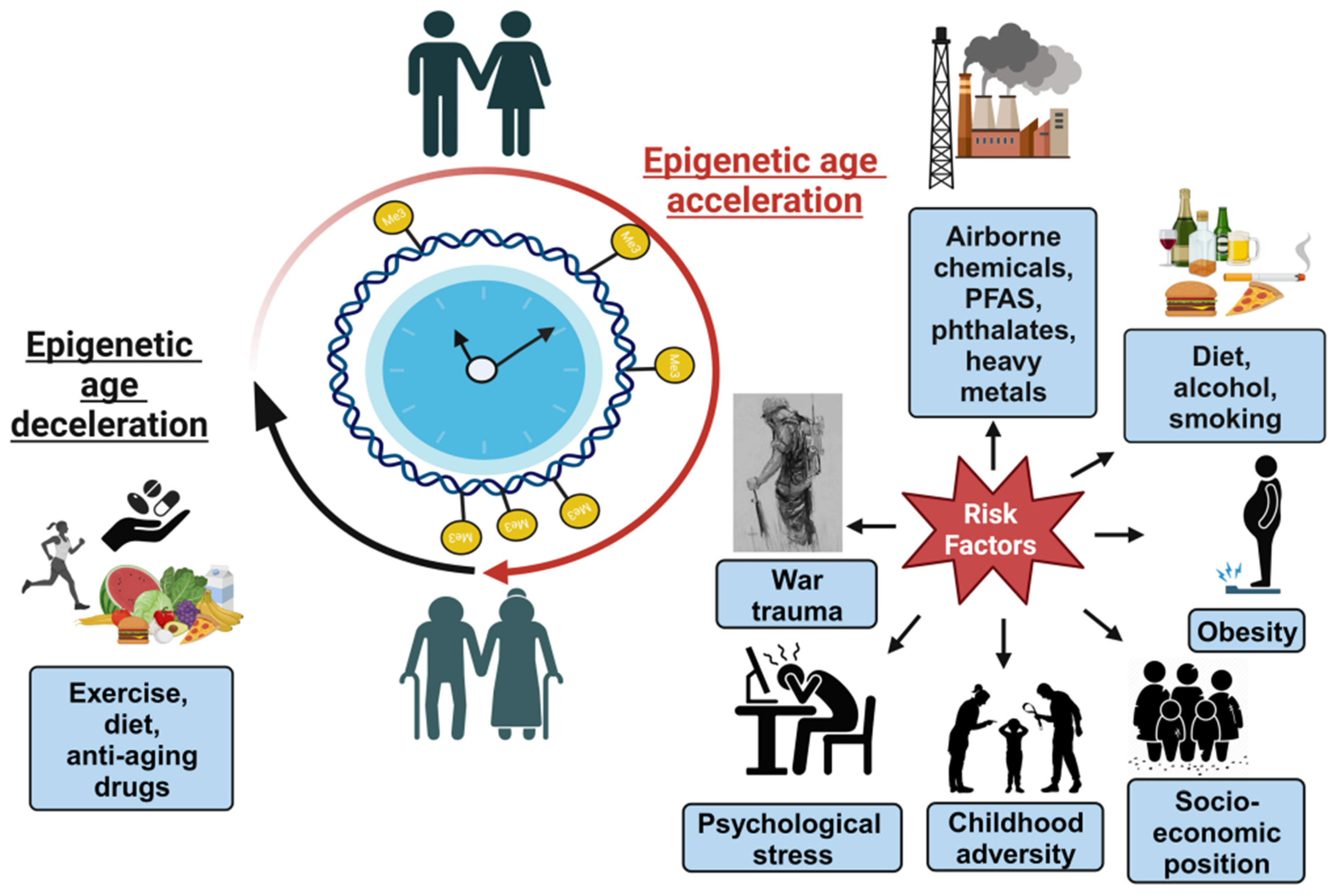
3. Part 2. Environmental Toxicants Affecting Epigenetic Clocks
3.1. Airborne Chemicals
3.2. Phthalates
3.3. Heavy Metals
3.4. Per- and Polyfluoroalkyl Substances
3.5. Psychosocial and Lifestyle Factors Affecting Epigenetic Clocks
3.6. Obesity and Its Effect on Epigenetic Clocks
3.7. Psychosocial Factors Affecting Newborn Epigenetic Aging
4. Discussion
5. Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khan, S.S.; Singer, B.D.; Vaughan, D.E. Molecular and physiological manifestations and measurement of aging in humans. Aging Cell 2017, 16, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Borkan, G.A.; Norris, A.H. Assessment of biological age using a profile of physical parameters. J. Gerontol. 1980, 35, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Ravindrarajah, R.; Lee, D.M.; Pye, S.R.; Gielen, E.; Boonen, S.; Vanderschueren, D.; Pendleton, N.; Finn, J.D.; Tajar, A.; O’Connell, M.D.; et al. European Male Aging Study, The ability of three different models of frailty to predict all-cause mortality: Results from the European Male Aging Study (EMAS). Arch. Gerontol. Geriatr. 2013, 57, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jockel, K.H.; Erbel, R.; Muhleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef] [PubMed]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Chen, B.H.; Colicino, E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Redmond, P.; Cox, S.R.; et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015, 16, 25. [Google Scholar] [CrossRef] [PubMed]
- Rae, M.J.; Butler, R.N.; Campisi, J.; de Grey, A.D.; Finch, C.E.; Gough, M.; Martin, G.M.; Vijg, J.; Perrott, K.M.; Logan, B.J. The demographic and biomedical case for late-life interventions in aging. Sci. Transl. Med. 2010, 2, 40cm21. [Google Scholar] [CrossRef] [PubMed]
- Harper, S. Economic and social implications of aging societies. Science 2014, 346, 587–591. [Google Scholar] [CrossRef]
- United Nations Department of Economic and Social Affairs, World Population Prospects; 2012 Revision, June 2013. Available online: https://population.un.org/wpp/ (accessed on 10 December 2023).
- Searle, S.D.; Mitnitski, A.; Gahbauer, E.A.; Gill, T.M.; Rockwood, K. A standard procedure for creating a frailty index. BMC Geriatr. 2008, 8, 24. [Google Scholar] [CrossRef]
- Baker, G.T., 3rd; Sprott, R.L. Biomarkers of aging. Exp. Gerontol. 1988, 23, 223–239. [Google Scholar] [CrossRef]
- Warner, H.R. Current status of efforts to measure and modulate the biological rate of aging. J. Gerontol. A Biol. Sci. Med. Sci. 2004, 59, 692–696. [Google Scholar] [CrossRef]
- Johnson, T.E. Recent results: Biomarkers of aging. Exp. Gerontol. 2006, 41, 1243–1246. [Google Scholar] [CrossRef] [PubMed]
- Butler, R.N.; Sprott, R.; Warner, H.; Bland, J.; Feuers, R.; Forster, M.; Fillit, H.; Harman, S.M.; Hewitt, M.; Hyman, M.; et al. Biomarkers of aging: From primitive organisms to humans. J. Gerontol. A Biol. Sci. Med. Sci. 2004, 59, B560–B567. [Google Scholar] [CrossRef] [PubMed]
- Mitnitski, A.; Collerton, J.; Martin-Ruiz, C.; Jagger, C.; von Zglinicki, T.; Rockwood, K.; Kirkwood, T.B. Age-related frailty and its association with biological markers of ageing. BMC Med. 2015, 13, 161. [Google Scholar] [CrossRef] [PubMed]
- Jylhava, J.; Pedersen, N.L.; Hagg, S. Biological Age Predictors. EBioMedicine 2017, 21, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Safaee, M.M.; Dwaraka, V.B.; Lee, J.M.; Fury, M.; Mendez, T.L.; Smith, R.; Lin, J.; Smith, D.L.; Burke, J.F.; Scheer, J.K.; et al. Epigenetic age biomarkers and risk assessment in adult spinal deformity: A novel association of biological age with frailty and disability. J. Neurosurg. Spine 2023, 38, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Franzago, M.; Pilenzi, L.; Di Rado, S.; Vitacolonna, E.; Stuppia, L. The epigenetic aging, obesity, and lifestyle. Front. Cell Dev. Biol. 2022, 10, 985274. [Google Scholar] [CrossRef]
- Moher, D.; Shamseer, L.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A.; Group, P.-P. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst. Rev. 2015, 4, 1. [Google Scholar] [CrossRef]
- Vetter, V.M.; Meyer, A.; Karbasiyan, M.; Steinhagen-Thiessen, E.; Hopfenmuller, W.; Demuth, I. Epigenetic Clock and Relative Telomere Length Represent Largely Different Aspects of Aging in the Berlin Aging Study II (BASE-II). J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 27–32. [Google Scholar] [CrossRef]
- Field, A.E.; Robertson, N.A.; Wang, T.; Havas, A.; Ideker, T.; Adams, P.D. DNA Methylation Clocks in Aging: Categories, Causes, and Consequences. Mol. Cell 2018, 71, 882–895. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115, Erratum in Genome Biol. 2015, 16, 96. [Google Scholar] [CrossRef] [PubMed]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.J.; Goodman, S.J.; Kobor, M.S. DNA methylation and healthy human aging. Aging Cell 2015, 14, 924–932. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.A.; Akman, K.; Calimport, S.R.; Wuttke, D.; Stolzing, A.; de Magalhaes, J.P. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res. 2012, 15, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Yi, S.V. Impacts of Chromatin States and Long-Range Genomic Segments on Aging and DNA Methylation. PLoS ONE 2015, 10, e0128517. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.F.; Tejedor, J.R.; Fernandez, A.F.; Fraga, M.F. Aging and cancer epigenetics: Where do the paths fork? Aging Cell 2022, 21, e13709. [Google Scholar] [CrossRef] [PubMed]
- Fox, B.C.; Mobley, H.L.; Wade, J.C. The use of a DNA probe for epidemiological studies of candidiasis in immunocompromised hosts. J. Infect. Dis. 1989, 159, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Ritchie, S.J.; Muniz-Terrera, G.; Harris, S.E.; Gibson, J.; Redmond, P.; Cox, S.R.; Pattie, A.; et al. The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int. J. Epidemiol. 2015, 44, 1388–1396. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef]
- Belsky, D.W.; Caspi, A.; Arseneault, L.; Baccarelli, A.; Corcoran, D.L.; Gao, X.; Hannon, E.; Harrington, H.L.; Rasmussen, L.J.; Houts, R.; et al. Quantification of the pace of biological aging in humans through a blood test, the DunedinPoAm DNA methylation algorithm. Elife 2020, 9, e54870. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wong, A.; Kuh, D.; Paul, D.S.; Rakyan, V.K.; Leslie, R.D.; Zheng, S.C.; Widschwendter, M.; Beck, S.; Teschendorff, A.E. Correlation of an epigenetic mitotic clock with cancer risk. Genome Biol. 2016, 17, 205. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E. A comparison of epigenetic mitotic-like clocks for cancer risk prediction. Genome Med. 2020, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Pilsner, J.R.; Saddiki, H.; Whitcomb, B.W.; Suvorov, A.; Louis, G.M.B.; Mumford, S.L.; Schisterman, E.F.; Oluwayiose, O.A.; Balzer, L.B. Sperm epigenetic clock associates with pregnancy outcomes in the general population. Hum. Reprod. 2022, 37, 1581–1593. [Google Scholar] [PubMed]
- Ren, X.; Kuan, P.F. RNAAgeCalc: A multi-tissue transcriptional age calculator. PLoS ONE 2020, 15, e0237006. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Hodar, J.A.; Del Sol, A. Measuring biological age using a functionally interpretable multi-tissue RNA clock. Aging Cell 2023, 22, e13799. [Google Scholar] [CrossRef] [PubMed]
- Trapp, A.; Kerepesi, C.; Gladyshev, V.N. Profiling epigenetic age in single cells. Nat. Aging 2021, 1, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Knight, A.K.; Craig, J.M.; Theda, C.; Baekvad-Hansen, M.; Bybjerg-Grauholm, J.; Hansen, C.S.; Hollegaard, M.V.; Hougaard, D.M.; Mortensen, P.B.; Weinsheimer, S.M.; et al. An epigenetic clock for gestational age at birth based on blood methylation data. Genome Biol. 2016, 17, 206. [Google Scholar] [CrossRef]
- Bohlin, J.; Haberg, S.E.; Magnus, P.; Reese, S.E.; Gjessing, H.K.; Magnus, M.C.; Parr, C.L.; Page, C.M.; London, S.J.; Nystad, W. Prediction of gestational age based on genome-wide differentially methylated regions. Genome Biol. 2016, 17, 207. [Google Scholar] [CrossRef]
- Lee, Y.; Choufani, S.; Weksberg, R.; Wilson, S.L.; Yuan, V.; Burt, A.; Marsit, C.; Lu, A.T.; Ritz, B.; Bohlin, J.; et al. Placental epigenetic clocks: Estimating gestational age using placental DNA methylation levels. Aging 2019, 11, 4238–4253. [Google Scholar] [CrossRef]
- McEwen, L.M.; O’Donnell, K.J.; McGill, M.G.; Edgar, R.D.; Jones, M.J.; MacIsaac, J.L.; Lin, D.T.S.; Ramadori, K.; Morin, A.; Gladish, N.; et al. The PedBE clock accurately estimates DNA methylation age in pediatric buccal cells. Proc. Natl. Acad. Sci. USA 2020, 117, 23329–23335. [Google Scholar] [CrossRef] [PubMed]
- Mayne, B.T.; Leemaqz, S.Y.; Smith, A.K.; Breen, J.; Roberts, C.T.; Bianco-Miotto, T. Accelerated placental aging in early onset preeclampsia pregnancies identified by DNA methylation. Epigenomics 2017, 9, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Graw, S.; Camerota, M.; Carter, B.S.; Helderman, J.; Hofheimer, J.A.; McGowan, E.C.; Neal, C.R.; Pastyrnak, S.L.; Smith, L.M.; DellaGrotta, S.A.; et al. NEOage clocks—Epigenetic clocks to estimate post-menstrual and postnatal age in preterm infants. Aging 2021, 13, 23527–23544. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Enroth, S.; Gyllensten, U. Continuous Aging of the Human DNA Methylome Throughout the Human Lifespan. PLoS ONE 2013, 8, e67378. [Google Scholar] [CrossRef] [PubMed]
- Florath, I.; Butterbach, K.; Muller, H.; Bewerunge-Hudler, M.; Brenner, H. Cross-sectional and longitudinal changes in DNA methylation with age: An epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum. Mol. Genet. 2014, 23, 1186–1201. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.E.; Shin, K.J.; Lee, H.Y. DNA methylation-based age prediction from various tissues and body fluids. BMB Rep. 2017, 50, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Maslau, S.; Andrew, T.; Yang, T.P.; Beyan, H.; Whittaker, P.; McCann, O.T.; Finer, S.; Valdes, A.M.; et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010, 20, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.T.; Tsai, P.C.; Yang, T.P.; Pidsley, R.; Nisbet, J.; Glass, D.; Mangino, M.; Zhai, G.; Zhang, F.; Valdes, A.; et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012, 8, e1002629. [Google Scholar] [CrossRef]
- Christensen, B.C.; Houseman, E.A.; Marsit, C.J.; Zheng, S.; Wrensch, M.R.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Padbury, J.F.; Bueno, R.; et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009, 5, e1000602. [Google Scholar] [CrossRef]
- Zheng, S.C.; Widschwendter, M.; Teschendorff, A.E. Epigenetic drift, epigenetic clocks and cancer risk. Epigenomics 2016, 8, 705–719. [Google Scholar] [CrossRef]
- Baranyi, G.; Deary, I.J.; McCartney, D.L.; Harris, S.E.; Shortt, N.; Reis, S.; Russ, T.C.; Thompson, C.W.; Vieno, M.; Cox, S.R.; et al. Life-course exposure to air pollution and biological ageing in the Lothian Birth Cohort 1936. Environ. Int. 2022, 169, 107501. [Google Scholar] [CrossRef] [PubMed]
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sanchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic predictor of age. PLoS ONE 2011, 6, e14821. [Google Scholar] [CrossRef] [PubMed]
- Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Gori, D.; Giuliani, C.; Mari, D.; Di Blasio, A.M.; Gentilini, D.; Vitale, G.; Collino, S.; et al. Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell 2012, 11, 1132–1134. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Mah, V.; Lu, A.T.; Woo, J.S.; Choi, O.W.; Jasinska, A.J.; Riancho, J.A.; Tung, S.; Coles, N.S.; Braun, J.; et al. The cerebellum ages slowly according to the epigenetic clock. Aging 2015, 7, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Arneson, A.; Haghani, A.; Thompson, M.J.; Pellegrini, M.; Kwon, S.B.; Vu, H.; Maciejewski, E.; Yao, M.; Li, C.Z.; Lu, A.T.; et al. A mammalian methylation array for profiling methylation levels at conserved sequences. Nat. Commun. 2022, 13, 783. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.T.; Fei, Z.; Haghani, A.; Robeck, T.R.; Zoller, J.A.; Li, C.Z.; Lowe, R.; Yan, Q.; Zhang, J.; Vu, H.; et al. Universal DNA Methylation Age across Mammalian Tissues. Nat Aging 2023, 3, 1144–1166. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Haghani, A.; Macoretta, N.; Ablaeva, J.; Zoller, J.A.; Li, C.Z.; Zhang, J.; Takasugi, M.; Zhao, Y.; Rydkina, E.; et al. DNA methylation clocks tick in naked mole rats but queens age more slowly than nonbreeders. Nat. Aging 2022, 2, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Prado, N.A.; Brown, J.L.; Zoller, J.A.; Haghani, A.; Yao, M.; Bagryanova, L.R.; Campana, M.G.; Maldonado, J.E.; Raj, K.; Schmitt, D.; et al. Epigenetic clock and methylation studies in elephants. Aging Cell 2021, 20, e13414. [Google Scholar] [CrossRef]
- Horvath, S.; Haghani, A.; Zoller, J.A.; Raj, K.; Sinha, I.; Robeck, T.R.; Black, P.; Couzens, A.; Lau, C.; Manoyan, M.; et al. Epigenetic clock and methylation studies in marsupials: Opossums, Tasmanian devils, kangaroos, and wallabies. Geroscience 2022, 44, 1825–1845. [Google Scholar] [CrossRef]
- Raj, K.; Szladovits, B.; Haghani, A.; Zoller, J.A.; Li, C.Z.; Black, P.; Maddox, D.; Robeck, T.R.; Horvath, S. Epigenetic clock and methylation studies in cats. Geroscience 2021, 43, 2363–2378. [Google Scholar] [CrossRef]
- Schachtschneider, K.M.; Schook, L.B.; Meudt, J.J.; Shanmuganayagam, D.; Zoller, J.A.; Haghani, A.; Li, C.Z.; Zhang, J.; Yang, A.; Raj, K.; et al. Epigenetic clock and DNA methylation analysis of porcine models of aging and obesity. Geroscience 2021, 43, 2467–2483. [Google Scholar] [CrossRef] [PubMed]
- Caulton, A.; Dodds, K.G.; McRae, K.M.; Couldrey, C.; Horvath, S.; Clarke, S.M. Development of Epigenetic Clocks for Key Ruminant Species. Genes 2021, 13, 96. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Haghani, A.; Peng, S.; Hales, E.N.; Zoller, J.A.; Raj, K.; Larison, B.; Robeck, T.R.; Petersen, J.L.; Bellone, R.R.; et al. DNA methylation aging and transcriptomic studies in horses. Nat. Commun. 2022, 13, 40. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.J.; Chwialkowska, K.; Rubbi, L.; Lusis, A.J.; Davis, R.C.; Srivastava, A.; Korstanje, R.; Churchill, G.A.; Horvath, S.; Pellegrini, M. A multi-tissue full lifespan epigenetic clock for mice. Aging 2018, 10, 2832–2854. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef]
- Danial, C.; Sarin, K.Y.; Oro, A.E.; Chang, A.L. An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib. Clin. Cancer Res. 2016, 22, 1325–1329. [Google Scholar] [CrossRef]
- Simpkin, A.J.; Howe, L.D.; Tilling, K.; Gaunt, T.R.; Lyttleton, O.; McArdle, W.L.; Ring, S.M.; Horvath, S.; Smith, G.D.; Relton, C.L. The epigenetic clock and physical development during childhood and adolescence: Longitudinal analysis from a UK birth cohort. Int. J. Epidemiol. 2017, 46, 549–558. [Google Scholar] [CrossRef]
- Chen, B.H.; Marioni, R.E.; Colicino, E.; Peters, M.J.; Ward-Caviness, C.K.; Tsai, P.C.; Roetker, N.S.; Just, A.C.; Demerath, E.W.; Guan, W.; et al. DNA methylation-based measures of biological age: Meta-analysis predicting time to death. Aging 2016, 8, 1844–1865. [Google Scholar] [CrossRef]
- Ignjatovic, V.; Lai, C.; Summerhayes, R.; Mathesius, U.; Tawfilis, S.; Perugini, M.A.; Monagle, P. Age-related differences in plasma proteins: How plasma proteins change from neonates to adults. PLoS ONE 2011, 6, e17213. [Google Scholar] [CrossRef]
- Ridker, P.M.; Buring, J.E.; Cook, N.R.; Rifai, N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: An 8-year follow-up of 14,719 initially healthy American women. Circulation 2003, 107, 391–397. [Google Scholar] [CrossRef]
- Belsky, D.W.; Caspi, A.; Corcoran, D.L.; Sugden, K.; Poulton, R.; Arseneault, L.; Baccarelli, A.; Chamarti, K.; Gao, X.; Hannon, E.; et al. DunedinPACE, a DNA methylation biomarker of the pace of aging. Elife 2022, 11, e73420. [Google Scholar] [CrossRef] [PubMed]
- Youn, A.; Wang, S. The MiAge Calculator: A DNA methylation-based mitotic age calculator of human tissue types. Epigenetics 2018, 13, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Kishino, T.; Takahashi, T.; Shimazu, T.; Charvat, H.; Kakugawa, Y.; Nakajima, T.; Lee, Y.C.; Iida, N.; Maeda, M.; et al. Genetic and epigenetic alterations in normal tissues have differential impacts on cancer risk among tissues. Proc. Natl. Acad. Sci. USA 2018, 115, 1328–1333. [Google Scholar] [CrossRef] [PubMed]
- Oluwayiose, O.A.; Wu, H.; Saddiki, H.; Whitcomb, B.W.; Balzer, L.B.; Brandon, N.; Suvorov, A.; Tayyab, R.; Sites, C.K.; Hill, L.; et al. Sperm DNA methylation mediates the association of male age on reproductive outcomes among couples undergoing infertility treatment. Sci. Rep. 2021, 11, 3216. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, T.G.; Aston, K.I.; Cairns, B.; Smith, A.; Carrell, D.T. Paternal germ line aging: DNA methylation age prediction from human sperm. BMC Genomics 2018, 19, 763. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA methylation aging clocks: Challenges and recommendations. Genome Biol. 2019, 20, 249. [Google Scholar] [CrossRef] [PubMed]
- Gravina, S.; Dong, X.; Yu, B.; Vijg, J. Single-cell genome-wide bisulfite sequencing uncovers extensive heterogeneity in the mouse liver methylome. Genome Biol. 2016, 17, 150. [Google Scholar] [CrossRef]
- de Magalhaes, J.P.; Curado, J.; Church, G.M. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics 2009, 25, 875–881. [Google Scholar] [CrossRef]
- Welle, S.; Brooks, A.I.; Delehanty, J.M.; Needler, N.; Thornton, C.A. Gene expression profile of aging in human muscle. Physiol. Genomics 2003, 14, 149–159. [Google Scholar] [CrossRef]
- Rodwell, G.E.; Sonu, R.; Zahn, J.M.; Lund, J.; Wilhelmy, J.; Wang, L.; Xiao, W.; Mindrinos, M.; Crane, E.; Segal, E.; et al. A transcriptional profile of aging in the human kidney. PLoS Biol. 2004, 2, e427. [Google Scholar] [CrossRef]
- Lu, T.; Pan, Y.; Kao, S.Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nature 2004, 429, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.; Vinuela, A.; Davies, M.N.; Ramasamy, A.; Parts, L.; Knowles, D.; Brown, A.A.; Hedman, A.K.; Small, K.S.; Buil, A.; et al. Gene expression changes with age in skin, adipose tissue, blood and brain. Genome Biol. 2013, 14, R75. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.J.; Joehanes, R.; Pilling, L.C.; Schurmann, C.; Conneely, K.N.; Powell, J.; Reinmaa, E.; Sutphin, G.L.; Zhernakova, A.; Schramm, K.; et al. The transcriptional landscape of age in human peripheral blood. Nat. Commun. 2015, 6, 8570. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, J.G.; Schulte, R.; Tsai, H.H.; Tyagi, S.; Ibarra, A.; Shokhirev, M.N.; Huang, L.; Hetzer, M.W.; Navlakha, S. Predicting age from the transcriptome of human dermal fibroblasts. Genome Biol. 2018, 19, 221. [Google Scholar] [CrossRef] [PubMed]
- GTex Consortium, The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [CrossRef] [PubMed]
- Hansen, J.; Meretzky, D.; Woldesenbet, S.; Stolovitzky, G.; Iyengar, R. A flexible ontology for inference of emergent whole cell function from relationships between subcellular processes. Sci. Rep. 2017, 7, 17689. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Zhou, L.; Perng, W.; Marsit, C.J.; Knight, A.K.; Cardenas, A.; Aung, M.T.; Hivert, M.F.; Aris, I.M.; Goodrich, J.M.; et al. Evaluation of pediatric epigenetic clocks across multiple tissues. Clin. Epigenetics 2023, 15, 142. [Google Scholar] [CrossRef] [PubMed]
- Heiss, C.; Spyridopoulos, I.; Haendeler, J. Interventions to slow cardiovascular aging: Dietary restriction, drugs and novel molecules. Exp. Gerontol. 2018, 109, 108–118. [Google Scholar] [CrossRef]
- Dato, S.; Bellizzi, D.; Rose, G.; Passarino, G. The impact of nutrients on the aging rate: A complex interaction of demographic, environmental and genetic factors. Mech. Ageing Dev. 2016, 154, 49–61. [Google Scholar] [CrossRef]
- Sae-Lee, C.; Corsi, S.; Barrow, T.M.; Kuhnle, G.G.C.; Bollati, V.; Mathers, J.C.; Byun, H.M. Dietary Intervention Modifies DNA Methylation Age Assessed by the Epigenetic Clock. Mol. Nutr. Food Res. 2018, 62, e1800092. [Google Scholar] [CrossRef]
- Monasso, G.S.; Kupers, L.K.; Jaddoe, V.W.V.; Heil, S.G.; Felix, J.F. Associations of circulating folate, vitamin B12 and homocysteine concentrations in early pregnancy and cord blood with epigenetic gestational age: The Generation R Study. Clin. Epigenetics 2021, 13, 95. [Google Scholar] [CrossRef] [PubMed]
- Martucci, M.; Ostan, R.; Biondi, F.; Bellavista, E.; Fabbri, C.; Bertarelli, C.; Salvioli, S.; Capri, M.; Franceschi, C.; Santoro, A. Mediterranean diet and inflammaging within the hormesis paradigm. Nutr. Rev. 2017, 75, 442–455. [Google Scholar] [CrossRef] [PubMed]
- Davinelli, S.; Trichopoulou, A.; Corbi, G.; De Vivo, I.; Scapagnini, G. The potential nutrigeroprotective role of Mediterranean diet and its functional components on telomere length dynamics. Ageing Res. Rev. 2019, 49, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gensous, N.; Garagnani, P.; Santoro, A.; Giuliani, C.; Ostan, R.; Fabbri, C.; Milazzo, M.; Gentilini, D.; di Blasio, A.M.; Pietruszka, B.; et al. One-year Mediterranean diet promotes epigenetic rejuvenation with country- and sex-specific effects: A pilot study from the NU-AGE project. Geroscience 2020, 42, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Fiorito, G.; Caini, S.; Palli, D.; Bendinelli, B.; Saieva, C.; Ermini, I.; Valentini, V.; Assedi, M.; Rizzolo, P.; Ambrogetti, D.; et al. DNA methylation-based biomarkers of aging were slowed down in a two-year diet and physical activity intervention trial: The DAMA study. Aging Cell 2021, 20, e13439. [Google Scholar] [CrossRef] [PubMed]
- Dugue, P.A.; Bassett, J.K.; Joo, J.E.; Baglietto, L.; Jung, C.H.; Wong, E.M.; Fiorito, G.; Schmidt, D.; Makalic, E.; Li, S.; et al. Association of DNA Methylation-Based Biological Age with Health Risk Factors and Overall and Cause-Specific Mortality. Am. J. Epidemiol. 2018, 187, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wong, E.M.; Nguyen, T.L.; Dite, G.S.; Stone, J.; Dugue, P.A.; Giles, G.G.; Southey, M.C.; Milne, R.L.; Hopper, J.L.; et al. DNA methylation-based biological age, genome-wide average DNA methylation, and conventional breast cancer risk factors. Sci. Rep. 2019, 9, 15055. [Google Scholar] [CrossRef]
- Galkin, F.; Kovalchuk, O.; Koldasbayeva, D.; Zhavoronkov, A.; Bischof, E. Stress; diet, exercise: Common environmental factors and their impact on epigenetic age. Ageing Res. Rev. 2023, 88, 101956. [Google Scholar] [CrossRef]
- Oblak, L.; van der Zaag, J.; Higgins-Chen, A.T.; Levine, M.E.; Boks, M.P. A systematic review of biological, social and environmental factors associated with epigenetic clock acceleration. Ageing Res. Rev. 2021, 69, 101348. [Google Scholar] [CrossRef]
- World Health Organization (Ed.) WHO Global Air Quality Guidelines: Particulate Matter (PM(2.5) and PM(10)), Ozone, Nitrogen Dioxide, Sulfur Dioxide and Carbon Monoxide; World Health Organization: Geneva, Switzerland, 2021.
- WHO. WHO Global Urban Ambient Air Pollution 2016 Database; WHO: Geneva, Switzerland, 2016.
- Alfano, R.; Herceg, Z.; Nawrot, T.S.; Chadeau-Hyam, M.; Ghantous, A.; Plusquin, M. The Impact of Air Pollution on Our Epigenome: How Far Is the Evidence? (A Systematic Review). Curr. Environ. Health Rep. 2018, 5, 544–578. [Google Scholar] [CrossRef]
- Ward-Caviness, C.K.; Nwanaji-Enwerem, J.C.; Wolf, K.; Wahl, S.; Colicino, E.; Trevisi, L.; Kloog, I.; Just, A.C.; Vokonas, P.; Cyrys, J.; et al. Long-term exposure to air pollution is associated with biological aging. Oncotarget 2016, 7, 74510–74525. [Google Scholar] [CrossRef] [PubMed]
- Nwanaji-Enwerem, J.C.; Dai, L.; Colicino, E.; Oulhote, Y.; Di, Q.; Kloog, I.; Just, A.C.; Hou, L.; Vokonas, P.; Baccarelli, A.A.; et al. Associations between long-term exposure to PM(2.5) component species and blood DNA methylation age in the elderly: The VA normative aging study. Environ. Int. 2017, 102, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Zhang, Y.; Li, G.; Sang, N. Potential Role of Inflammation in Associations between Particulate Matter and Heart Failure. Curr. Pharm. Des. 2018, 24, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Cheng, Z.; Li, N.; Mao, S.; Ma, R.; He, H.; Niu, Z.; Chen, X.; Xiang, H. The short- and long-term associations of particulate matter with inflammation and blood coagulation markers: A meta-analysis. Environ. Pollut. 2020, 267, 115630. [Google Scholar] [CrossRef] [PubMed]
- Bell, M.L.; Dominici, F.; Ebisu, K.; Zeger, S.L.; Samet, J.M. Spatial and temporal variation in PM(2.5) chemical composition in the United States for health effects studies. Environ. Health Perspect. 2007, 115, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Cantone, L.; Tobaldini, E.; Favero, C.; Albetti, B.; Sacco, R.M.; Torgano, G.; Ferrari, L.; Montano, N.; Bollati, V. Particulate Air Pollution, Clock Gene Methylation, and Stroke: Effects on Stroke Severity and Disability. Int. J. Mol. Sci. 2020, 21, 3090. [Google Scholar] [CrossRef] [PubMed]
- Nawrot, T.S.; Saenen, N.D.; Schenk, J.; Janssen, B.G.; Motta, V.; Tarantini, L.; Cox, B.; Lefebvre, W.; Vanpoucke, C.; Maggioni, C.; et al. Placental circadian pathway methylation and in utero exposure to fine particle air pollution. Environ. Int. 2018, 114, 231–241. [Google Scholar] [CrossRef]
- Shi, W.; Tang, S.; Fang, J.; Cao, Y.; Chen, C.; Li, T.; Gao, X.; Shi, X. Epigenetic age stratifies the risk of blood pressure elevation related to short-term PM(2.5) exposure in older adults. Environ. Res. 2022, 212, 113507. [Google Scholar] [CrossRef]
- van der Laan, L.; Cardenas, A.; Vermeulen, R.; Fadadu, R.P.; Hubbard, A.E.; Phillips, R.V.; Zhang, L.; Breeze, C.; Hu, W.; Wen, C.; et al. Epigenetic aging biomarkers and occupational exposure to benzene, trichloroethylene and formaldehyde. Environ. Int. 2022, 158, 106871. [Google Scholar] [CrossRef]
- Taylor, A.M.; Pattie, A.; Deary, I.J. Cohort Profile Update: The Lothian Birth Cohorts of 1921 and 1936. Int. J. Epidemiol. 2018, 47, 1042–1042r. [Google Scholar] [CrossRef]
- de Prado-Bert, P.; Ruiz-Arenas, C.; Vives-Usano, M.; Andrusaityte, S.; Cadiou, S.; Carracedo, A.; Casas, M.; Chatzi, L.; Dadvand, P.; Gonzalez, J.R.; et al. The early-life exposome and epigenetic age acceleration in children. Environ. Int. 2021, 155, 106683. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.O. Environment, susceptibility windows, development, and child health. Curr. Opin. Pediatr. 2017, 29, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Song, A.Y.; Feinberg, J.I.; Bakulski, K.M.; Croen, L.A.; Fallin, M.D.; Newschaffer, C.J.; Hertz-Picciotto, I.; Schmidt, R.J.; Ladd-Acosta, C.; Volk, H.E. Prenatal Exposure to Ambient Air Pollution and Epigenetic Aging at Birth in Newborns. Front. Genet. 2022, 13, 929416. [Google Scholar] [CrossRef]
- Dutta, S.; Haggerty, D.K.; Rappolee, D.A.; Ruden, D.M. Phthalate Exposure and Long-Term Epigenomic Consequences: A Review. Front. Genet. 2020, 11, 405. [Google Scholar] [CrossRef] [PubMed]
- Sheng, N.; Wang, J.; Xing, F.; Duan, X.; Xiang, Z. Associations between exposure to phthalates and rheumatoid arthritis risk among adults in NHANES, 2007–2016. Chemosphere 2023, 338, 139472. [Google Scholar] [CrossRef]
- Khodasevich, D.; Holland, N.; Hubbard, A.; Harley, K.; Deardorff, J.; Eskenazi, B.; Cardenas, A. Associations between prenatal phthalate exposure and childhood epigenetic age acceleration. Environ. Res. 2023, 231, 116067. [Google Scholar] [CrossRef]
- Han, Q.; Gao, X.; Wang, S.; Wei, Z.; Wang, Y.; Xu, K.; Chen, M. Co-exposure to polystyrene microplastics and di-(2-ethylhexyl) phthalate aggravates allergic asthma through the TRPA1-p38 MAPK pathway. Toxicol. Lett. 2023, 384, 73–85. [Google Scholar] [CrossRef]
- Shi, W.; Gao, X.; Cao, Y.; Chen, Y.; Cui, Q.; Deng, F.; Yang, B.; Lin, E.Z.; Fang, J.; Li, T.; et al. Personal airborne chemical exposure and epigenetic ageing biomarkers in healthy Chinese elderly individuals: Evidence from mixture approaches. Environ. Int. 2022, 170, 107614. [Google Scholar] [CrossRef]
- Oluwayiose, O.A.; Houle, E.; Wu, H.; Whitcomb, B.W.; Mumford, S.L.; Schisterman, E.F.; Suvorov, A.; Balzer, L.B.; Pilsner, J.R. Urinary phthalate metabolites and their mixtures are associated with advanced sperm epigenetic aging in a general population. Environ. Res. 2022, 214, 114115. [Google Scholar] [CrossRef]
- Wu, H.; Estill, M.S.; Shershebnev, A.; Suvorov, A.; Krawetz, S.A.; Whitcomb, B.W.; Dinnie, H.; Rahil, T.; Sites, C.K.; Pilsner, J.R. Preconception urinary phthalate concentrations and sperm DNA methylation profiles among men undergoing IVF treatment: A cross-sectional study. Hum. Reprod. 2017, 32, 2159–2169. [Google Scholar] [CrossRef]
- Wang, Y.; Karlsson, R.; Jylhava, J.; Hedman, A.K.; Almqvist, C.; Karlsson, I.K.; Pedersen, N.L.; Almgren, M.; Hagg, S. Comprehensive longitudinal study of epigenetic mutations in aging. Clin. Epigenetics 2019, 11, 187. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of Iron Homeostasis: New Players in Metabolism, Cell Death, and Disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Sfera, A.; Bullock, K.; Price, A.; Inderias, L.; Osorio, C. Ferrosenescence: The iron age of neurodegeneration? Mech. Ageing Dev. 2018, 174, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.T.; Haupt, S.; Haupt, Y.; Denoyer, D.; Adlard, P.A.; Bush, A.I.; et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018, 14, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Mangan, D. Iron: An underrated factor in aging. Aging 2021, 13, 23407–23415. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, D.; Hu, Y. Appraising the Associations Between Systemic Iron Status and Epigenetic Clocks: A Genetic Correlation and Bidirectional Mendelian Randomization Study. Am. J. Clin. Nutr. 2023, 118, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Boyer, K.; Domingo-Relloso, A.; Jiang, E.; Haack, K.; Goessler, W.; Zhang, Y.; Umans, J.G.; Belsky, D.W.; Cole, S.A.; Navas-Acien, A.; et al. Metal mixtures and DNA methylation measures of biological aging in American Indian populations. Environ. Int. 2023, 178, 108064. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Peng, R.D.; Jones, M.R.; Francesconi, K.A.; Goessler, W.; Howard, B.V.; Umans, J.G.; Best, L.G.; Guallar, E.; Post, W.S.; et al. Metal mixtures in urban and rural populations in the US: The Multi-Ethnic Study of Atherosclerosis and the Strong Heart Study. Environ. Res. 2016, 147, 356–364. [Google Scholar] [CrossRef]
- Galvez-Fernandez, M.; Powers, M.; Grau-Perez, M.; Domingo-Relloso, A.; Lolacono, N.; Goessler, W.; Zhang, Y.; Fretts, A.; Umans, J.G.; Maruthur, N.; et al. Urinary Zinc and Incident Type 2 Diabetes: Prospective Evidence from the Strong Heart Study. Diabetes Care 2022, 45, 2561–2569. [Google Scholar] [CrossRef]
- Lodge, E.K.; Dhingra, R.; Martin, C.L.; Fry, R.C.; White, A.J.; Ward-Caviness, C.K.; Wani, A.H.; Uddin, M.; Wildman, D.E.; Galea, S.; et al. Serum lead, mercury, manganese, and copper and DNA methylation age among adults in Detroit, Michigan. Environ. Epigenetics 2022, 8, dvac018. [Google Scholar] [CrossRef]
- Evich, M.G.; Davis, M.J.B.; McCord, J.P.; Acrey, B.; Awkerman, J.A.; Knappe, D.R.U.; Lindstrom, A.B.; Speth, T.F.; Tebes-Stevens, C.; Strynar, M.J.; et al. Per- and polyfluoroalkyl substances in the environment. Science 2022, 375, eabg9065. [Google Scholar] [CrossRef] [PubMed]
- Sunderland, E.M.; Hu, X.C.; Dassuncao, C.; Tokranov, A.K.; Wagner, C.C.; Allen, J.G. A review of the pathways of human exposure to poly- and perfluoroalkyl substances (PFASs) and present understanding of health effects. J. Expo. Sci. Environ. Epidemiol. 2019, 29, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Schulz, K.; Silva, M.R.; Klaper, R. Distribution and effects of branched versus linear isomers of PFOA, PFOS, and PFHxS: A review of recent literature. Sci. Total Environ. 2020, 733, 139186. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Eliot, M.N.; Papandonatos, G.D.; Kelsey, K.T.; Fore, R.; Langevin, S.; Buckley, J.; Chen, A.; Lanphear, B.P.; Cecil, K.M.; et al. Gestational Perfluoroalkyl Substance Exposure and DNA Methylation at Birth and 12 Years of Age: A Longitudinal Epigenome-Wide Association Study. Environ. Health Perspect. 2022, 130, 37005. [Google Scholar] [CrossRef] [PubMed]
- Starling, A.P.; Liu, C.; Shen, G.; Yang, I.V.; Kechris, K.; Borengasser, S.J.; Boyle, K.E.; Zhang, W.; Smith, H.A.; Calafat, A.M.; et al. Prenatal Exposure to Per- and Polyfluoroalkyl Substances, Umbilical Cord Blood DNA Methylation, and Cardio-Metabolic Indicators in Newborns: The Healthy Start Study. Environ. Health Perspect. 2020, 128, 127014. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jurkovic-Mlakar, S.; Lindh, C.H.; Scott, K.; Fletcher, T.; Jakobsson, K.; Engstrom, K. Associations between serum concentrations of perfluoroalkyl substances and DNA methylation in women exposed through drinking water: A pilot study in Ronneby, Sweden. Environ. Int. 2020, 145, 106148. [Google Scholar] [CrossRef]
- Xu, Y.; Lindh, C.H.; Fletcher, T.; Jakobsson, K.; Engstrom, K. Perfluoroalkyl substances influence DNA methylation in school-age children highly exposed through drinking water contaminated from firefighting foam: A cohort study in Ronneby, Sweden. Environ. Epigenet. 2022, 8, dvac004. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.M.; Calkins, M.M.; Caban-Martinez, A.J.; Stueckle, T.; Grant, C.; Calafat, A.M.; Nematollahi, A.; Jung, A.M.; Graber, J.M.; Jenkins, T.; et al. Per- and polyfluoroalkyl substances, epigenetic age and DNA methylation: A cross-sectional study of firefighters. Epigenomics 2021, 13, 1619–1636. [Google Scholar] [CrossRef]
- Epel, E.S.; Blackburn, E.H.; Lin, J.; Dhabhar, F.S.; Adler, N.E.; Morrow, J.D.; Cawthon, R.M. Accelerated telomere shortening in response to life stress. Proc. Natl. Acad. Sci. USA 2004, 101, 17312–17315. [Google Scholar] [CrossRef]
- Hershey, H.P.; Barker, R.F.; Idler, K.B.; Murray, M.G.; Quail, P.H. Nucleotide sequence and characterization of a gene encoding the phytochrome polypeptide from Avena. Gene 1987, 61, 339–348. [Google Scholar] [CrossRef]
- Fries, G.R.; Bauer, I.E.; Scaini, G.; Wu, M.J.; Kazimi, I.F.; Valvassori, S.S.; Zunta-Soares, G.; Walss-Bass, C.; Soares, J.C.; Quevedo, J. Accelerated epigenetic aging and mitochondrial DNA copy number in bipolar disorder. Transl. Psychiatry 2017, 7, 1283. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.J.; Logue, M.W.; Hayes, J.P.; Sadeh, N.; Schichman, S.A.; Stone, A.; Salat, D.H.; Milberg, W.; McGlinchey, R.; Miller, M.W. Accelerated DNA methylation age: Associations with PTSD and neural integrity. Psychoneuroendocrinology 2016, 63, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W.; Sadeh, N. Traumatic stress, oxidative stress and post-traumatic stress disorder: Neurodegeneration and the accelerated-aging hypothesis. Mol. Psychiatry 2014, 19, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Kiecolt-Glaser, J.K.; Wilson, S.J.; Disorders, P. Morbidity, and Mortality: Tracing Mechanistic Pathways to Accelerated Aging. Psychosom. Med. 2016, 78, 772–775. [Google Scholar] [CrossRef] [PubMed]
- Darrow, S.M.; Verhoeven, J.E.; Revesz, D.; Lindqvist, D.; Penninx, B.W.; Delucchi, K.L.; Wolkowitz, O.M.; Mathews, C.A. The Association between Psychiatric Disorders and Telomere Length: A Meta-Analysis Involving 14,827 Persons. Psychosom. Med. 2016, 78, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Protsenko, E.; Wolkowitz, O.M.; Yaffe, K. Associations of stress and stress-related psychiatric disorders with GrimAge acceleration: Review and suggestions for future work. Transl. Psychiatry 2023, 13, 142. [Google Scholar] [CrossRef] [PubMed]
- Tollenaar, M.S.; Beijers, R.; Garg, E.; Nguyen, T.T.T.; Lin, D.T.S.; MacIsaac, J.L.; Shalev, I.; Kobor, M.S.; Meaney, M.J.; O’Donnell, K.J.; et al. Internalizing symptoms associate with the pace of epigenetic aging in childhood. Biol. Psychol. 2021, 159, 108021. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.J.; Logue, M.W.; Stoop, T.B.; Schichman, S.A.; Stone, A.; Sadeh, N.; Hayes, J.P.; Miller, M.W. Accelerated DNA Methylation Age: Associations with Posttraumatic Stress Disorder and Mortality. Psychosom. Med. 2018, 80, 42–48. [Google Scholar] [CrossRef]
- Wolf, E.J.; Logue, M.W.; Morrison, F.G.; Wilcox, E.S.; Stone, A.; Schichman, S.A.; McGlinchey, R.E.; Milberg, W.P.; Miller, M.W. Posttraumatic psychopathology and the pace of the epigenetic clock: A longitudinal investigation. Psychol. Med. 2019, 49, 791–800. [Google Scholar] [CrossRef]
- Wolf, E.J.; Maniates, H.; Nugent, N.; Maihofer, A.X.; Armstrong, D.; Ratanatharathorn, A.; Ashley-Koch, A.E.; Garrett, M.; Kimbrel, N.A.; Lori, A.; et al. Traumatic stress and accelerated DNA methylation age: A meta-analysis. Psychoneuroendocrinology 2018, 92, 123–134. [Google Scholar] [CrossRef]
- Mehta, D.; Bruenig, D.; Pierce, J.; Sathyanarayanan, A.; Stringfellow, R.; Miller, O.; Mullens, A.B.; Shakespeare-Finch, J. Recalibrating the epigenetic clock after exposure to trauma: The role of risk and protective psychosocial factors. J. Psychiatr. Res. 2022, 149, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Harvanek, Z.M.; Fogelman, N.; Xu, K.; Sinha, R. Psychological and biological resilience modulates the effects of stress on epigenetic aging. Transl. Psychiatry 2021, 11, 601. [Google Scholar] [CrossRef] [PubMed]
- Pi-Sunyer, X. The medical risks of obesity. Postgrad. Med. 2009, 121, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Tam, B.T.; Morais, J.A.; Santosa, S. Obesity and ageing: Two sides of the same coin. Obes. Rev. 2020, 21, e12991. [Google Scholar] [CrossRef] [PubMed]
- Fraszczyk, E.; Luijten, M.; Spijkerman, A.M.W.; Snieder, H.; Wackers, P.F.K.; Bloks, V.W.; Nicoletti, C.F.; Nonino, C.B.; Crujeiras, A.B.; Buurman, W.A.; et al. The effects of bariatric surgery on clinical profile, DNA methylation, and ageing in severely obese patients. Clin. Epigenetics 2020, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Dammering, F.; Martins, J.; Dittrich, K.; Czamara, D.; Rex-Haffner, M.; Overfeld, J.; de Punder, K.; Buss, C.; Entringer, S.; Winter, S.M.; et al. The pediatric buccal epigenetic clock identifies significant ageing acceleration in children with internalizing disorder and maltreatment exposure. Neurobiol. Stress 2021, 15, 100394. [Google Scholar] [CrossRef]
- Quinn, E.B.; Hsiao, C.J.; Maisha, F.M.; Mulligan, C.J. Prenatal maternal stress is associated with site-specific and age acceleration changes in maternal and newborn DNA methylation. Epigenetics 2023, 18, 2222473. [Google Scholar] [CrossRef]
- Hamlat, E.J.; Prather, A.A.; Horvath, S.; Belsky, J.; Epel, E.S. Early life adversity; pubertal timing, and epigenetic age acceleration in adulthood. Dev. Psychobiol. 2021, 63, 890–902. [Google Scholar] [CrossRef]
- McGill, M.G.; Pokhvisneva, I.; Clappison, A.S.; McEwen, L.M.; Beijers, R.; Tollenaar, M.S.; Pham, H.; Kee, M.Z.L.; Garg, E.; de Mendonca Filho, E.J.; et al. Maternal Prenatal Anxiety and the Fetal Origins of Epigenetic Aging. Biol. Psychiatry 2022, 91, 303–312. [Google Scholar] [CrossRef]
- Shiau, S.; Wang, L.; Liu, H.; Zheng, Y.; Drong, A.; Joyce, B.T.; Wang, J.; Li, W.; Leng, J.; Shen, Y.; et al. Prenatal gestational diabetes mellitus exposure and accelerated offspring DNA methylation age in early childhood. Epigenetics 2021, 16, 186–195. [Google Scholar] [CrossRef]
- Clark, J.; Bulka, C.M.; Martin, C.L.; Roell, K.; Santos, H.P.; O’Shea, T.M.; Smeester, L.; Fry, R.; Dhingra, R. Placental epigenetic gestational aging in relation to maternal sociodemographic factors and smoking among infants born extremely preterm: A descriptive study. Epigenetics 2022, 17, 2389–2403. [Google Scholar] [CrossRef] [PubMed]
- Katrinli, S.; Smith, A.K.; Drury, S.S.; Covault, J.; Ford, J.D.; Singh, V.; Reese, B.; Johnson, A.; Scranton, V.; Fall, P.; et al. Cumulative stress, PTSD, and emotion dysregulation during pregnancy and epigenetic age acceleration in Hispanic mothers and their newborn infants. Epigenetics 2023, 18, 2231722. [Google Scholar] [CrossRef] [PubMed]
- Haftorn, K.L.; Lee, Y.; Denault, W.R.P.; Page, C.M.; Nustad, H.E.; Lyle, R.; Gjessing, H.K.; Malmberg, A.; Magnus, M.C.; Naess, O.; et al. An EPIC predictor of gestational age and its application to newborns conceived by assisted reproductive technologies. Clin. Epigenetics 2021, 13, 82. [Google Scholar] [CrossRef] [PubMed]
- Dye, C.K.; Wu, H.; Monk, C.; Belsky, D.W.; Alschuler, D.; Lee, S.; O’Donnell, K.; Scorza, P. Mother’s childhood adversity is associated with accelerated epigenetic aging in pregnancy and in male newborns. bioRxiv 2023. [Google Scholar] [CrossRef]
- Koen, N.; Jones, M.J.; Nhapi, R.T.; Lake, M.T.; Donald, K.A.; Barnett, W.; Hoffman, N.; MacIsaac, J.L.; Morin, A.M.; Lin, D.T.S.; et al. Maternal psychosocial risk factors and child gestational epigenetic age in a South African birth cohort study. Transl. Psychiatry 2021, 11, 358. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.J.; Chandra, T. Epigenetic age prediction. Aging Cell 2021, 20, e13452. [Google Scholar] [CrossRef]
- Dupras, C.; Beck, S.; Rothstein, M.A.; Berner, A.; Saulnier, K.M.; Pinkesz, M.; Prince, A.E.R.; Liosi, S.; Song, L.; Joly, Y. Potential (mis)use of epigenetic age estimators by private companies and public agencies: Human rights law should provide ethical guidance. Environ. Epigenetics 2019, 5, dvz018. [Google Scholar]
- Tamatea, A.J.; Psychopathy, B. Legal, and Research Implications at the Interface of Epigenetics and Chronic Antisocial Conduct. Behav. Sci. Law. 2015, 33, 629–643. [Google Scholar] [CrossRef]
- Zannas, A.S.; Arloth, J.; Carrillo-Roa, T.; Iurato, S.; Roh, S.; Ressler, K.J.; Nemeroff, C.B.; Smith, A.K.; Bradley, B.; Heim, C.; et al. Lifetime stress accelerates epigenetic aging in an urban, African American cohort: Relevance of glucocorticoid signaling. Genome Biol. 2015, 16, 266. [Google Scholar] [CrossRef]
- Poganik, J.R.; Zhang, B.; Baht, G.S.; Tyshkovskiy, A.; Deik, A.; Kerepesi, C.; Yim, S.H.; Lu, A.T.; Haghani, A.; Gong, T.; et al. Biological age is increased by stress and restored upon recovery. Cell Metab. 2023, 35, 807–820.e5. [Google Scholar] [CrossRef]
- Wang, K.; Liu, H.; Hu, Q.; Wang, L.; Liu, J.; Zheng, Z.; Zhang, W.; Ren, J.; Zhu, F.; Liu, G.H. Epigenetic regulation of aging: Implications for interventions of aging and diseases. Signal Transduct. Target. Ther. 2022, 7, 374. [Google Scholar] [CrossRef] [PubMed]
- Sidler, C.; Kovalchuk, O.; Kovalchuk, I. Epigenetic Regulation of Cellular Senescence and Aging. Front. Genet. 2017, 8, 138. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Traldi, P.; Hamdan, M. Mass Spectrometry-Based Proteomics: Analyses Related to Drug-Resistance and Disease Biomarkers. Medicina 2023, 59, 1722. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Fu, Y.; Wang, X.; Wu, R.; Su, D.; Zhou, N.; Qi, Y. Metformin protects lens epithelial cells against senescence in a naturally aged mouse model. Cell Death Discov. 2022, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Thompson, J.; Hu, Y.; Lesnefsky, E.J. Chronic metformin treatment decreases cardiac injury during ischemia-reperfusion by attenuating endoplasmic reticulum stress with improved mitochondrial function. Aging 2021, 13, 7828–7845. [Google Scholar] [CrossRef] [PubMed]
- Strong, R.; Miller, R.A.; Bogue, M.; Fernandez, E.; Javors, M.A.; Libert, S.; Marinez, P.A.; Murphy, M.P.; Musi, N.; Nelson, J.F.; et al. Rapamycin-mediated mouse lifespan extension: Late-life dosage regimes with sex-specific effects. Aging Cell 2020, 19, e13269. [Google Scholar] [CrossRef]
- Quarles, E.; Basisty, N.; Chiao, Y.A.; Merrihew, G.; Gu, H.; Sweetwyne, M.T.; Fredrickson, J.; Nguyen, N.H.; Razumova, M.; Kooiker, K.; et al. Rapamycin persistently improves cardiac function in aged, male and female mice, even following cessation of treatment. Aging Cell 2020, 19, e13086. [Google Scholar] [CrossRef]
- Kawakami, Y.; Hambright, W.S.; Takayama, K.; Mu, X.; Lu, A.; Cummins, J.H.; Matsumoto, T.; Yurube, T.; Kuroda, R.; Kurosaka, M.; et al. Rapamycin Rescues Age-Related Changes in Muscle-Derived Stem/Progenitor Cells from Progeroid Mice. Mol. Ther. Methods Clin. Dev. 2019, 14, 64–76. [Google Scholar] [CrossRef]
- Garcia, D.N.; Saccon, T.D.; Pradiee, J.; Rincon, J.A.A.; Andrade, K.R.S.; Rovani, M.T.; Mondadori, R.G.; Cruz, L.A.X.; Barros, C.C.; Masternak, M.M.; et al. Effect of caloric restriction and rapamycin on ovarian aging in mice. Geroscience 2019, 41, 395–408. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Type of Clock | Name of the Clock | Publication Year and Ref. | # of CpG | No. of Subjects (N) | Age Range | Tissue | Training Phenotype | Platform | |
|---|---|---|---|---|---|---|---|---|---|
| DNAm -based molecular epigenetic clocks | G1 clocks | Horvath’s clock | 2013; [22] | 353 | 7844 | 0–100 | 51 healthy tissues & cell types | C-age | Illumina 27 K and 450 K |
| Hannum’s clock | 2013; [23] | 71 | 482 | 19–101 | Whole blood | C-age | 450 K | ||
| G2 clocks | DNAm PhenoAge | 2018; [30] | 513 | 9926 | 21–100 | Multiple | Lifespan (mortality risk score) | 27 K and 450 K and EPIC | |
| DNAm GrimAge | 2019; [31] | 1030 | 6935 | 46–78 | Whole blood | Lifespan (mortality risk score) | 450 K and EPIC | ||
| G3 Clocks | Dunedin PACE | 2020; [32] | 46 | 810 | 26–38 | Whole Blood | Pace of Aging | 450 K and EPIC | |
| DNAm- based mitotic clocks | EpiTOC | 2016; [33] | 385 | 656 | 19–101 | Whole blood | Mitotic age, cancer risk | 450 K | |
| MiAge | 2018; [33] | 286 | 4020 | N/A | 8 TTGA Cancer cells | Mitotic age, cancer risk, survival | 450 K | ||
| EpiTOC2 | 2020; [34] | 385 | 656 | 19–101 | Whole blood | Mitotic age, cancer risk | 450 K | ||
| Sperm epigenetic clocks | Sperm epigenetic aging (SEA)CpG clock | 2022; [35] | 803,063 | 379 semen samples | ≥18 | Semen | Sperm epigenetic aging | EPIC Infinium Methylation Beadchip | |
| RNA clocks | RNAAgeCalc | 2020; [36] | 353 | 9448 samples | N/A | Multi-tissue | Transcriptional age | RNA-Seq data from the Genotype-Tissue Expression (GTEx) Program | |
| MultiTIMER | 2023; [37] | N/A | 23,000 annotated samples | N/A | Multi-tissue | C-age | RNA-seq samples (ArchS4) v11 | ||
| Single-cell epigenetic clock framework (scAge) | scAge | 2021; [38] | 500,000 CpGs/ cell | 549 tissue samples | 1–21 months old mice | Multi-tissue | Single-cell B-age predictions | Computational platform | |
| Pediatric epigenetic clocks | Knight’s clock | 2016; [39] | 148 | 207 | 24–42 weeks | Cord blood | Gestational Age | 27 K and 450 K | |
| Bohlin’s clock | 2016; [40] | 132 | 685 | Neonates | Cord blood | Gestational Age | 450 K | ||
| Lee’s placental clock | 2019; [41] | 441,870 | 1102 | 5–42 weeks | Placenta | Gestational Age | 450 K and EPIC | ||
| Pediatric-Buccal-Epigenetic (PedBE) clock | 2020; [42] | 94 | 1032 | 0 to 20 years old | Buccal epithelial cels | Pediatric age | 450 K and EPIC | ||
| Mayne’s clock | 2017; [43] | 62 | 409 | 8–42 weeks | Placenta | Gestational age | 27 K and 450 K | ||
| NeoAge clocks (4 epigenetic clocks) | 2021; [44] | 303–522 | 542 | Pre-term infants (<30 weeks) | Buccal cell samples | Post-menstrual and postnatal age of neonates | 450 K and EPIC | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dutta, S.; Goodrich, J.M.; Dolinoy, D.C.; Ruden, D.M. Biological Aging Acceleration Due to Environmental Exposures: An Exciting New Direction in Toxicogenomics Research. Genes 2024, 15, 16. https://doi.org/10.3390/genes15010016
Dutta S, Goodrich JM, Dolinoy DC, Ruden DM. Biological Aging Acceleration Due to Environmental Exposures: An Exciting New Direction in Toxicogenomics Research. Genes. 2024; 15(1):16. https://doi.org/10.3390/genes15010016
Chicago/Turabian StyleDutta, Sudipta, Jaclyn M. Goodrich, Dana C. Dolinoy, and Douglas M. Ruden. 2024. "Biological Aging Acceleration Due to Environmental Exposures: An Exciting New Direction in Toxicogenomics Research" Genes 15, no. 1: 16. https://doi.org/10.3390/genes15010016
APA StyleDutta, S., Goodrich, J. M., Dolinoy, D. C., & Ruden, D. M. (2024). Biological Aging Acceleration Due to Environmental Exposures: An Exciting New Direction in Toxicogenomics Research. Genes, 15(1), 16. https://doi.org/10.3390/genes15010016