
Lessons Learned from Translating Genome Sequencing to Clinical Routine: Understanding the Accuracy of a Diagnostic Pipeline

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Diagnostic Exome and Genome Sequencing
2.2. Data Analysis Pipeline and Decision Support System
2.3. Pipeline Validation
2.4. Quality Control
2.5. Coverage Statistics
2.6. Variant Count Statistics
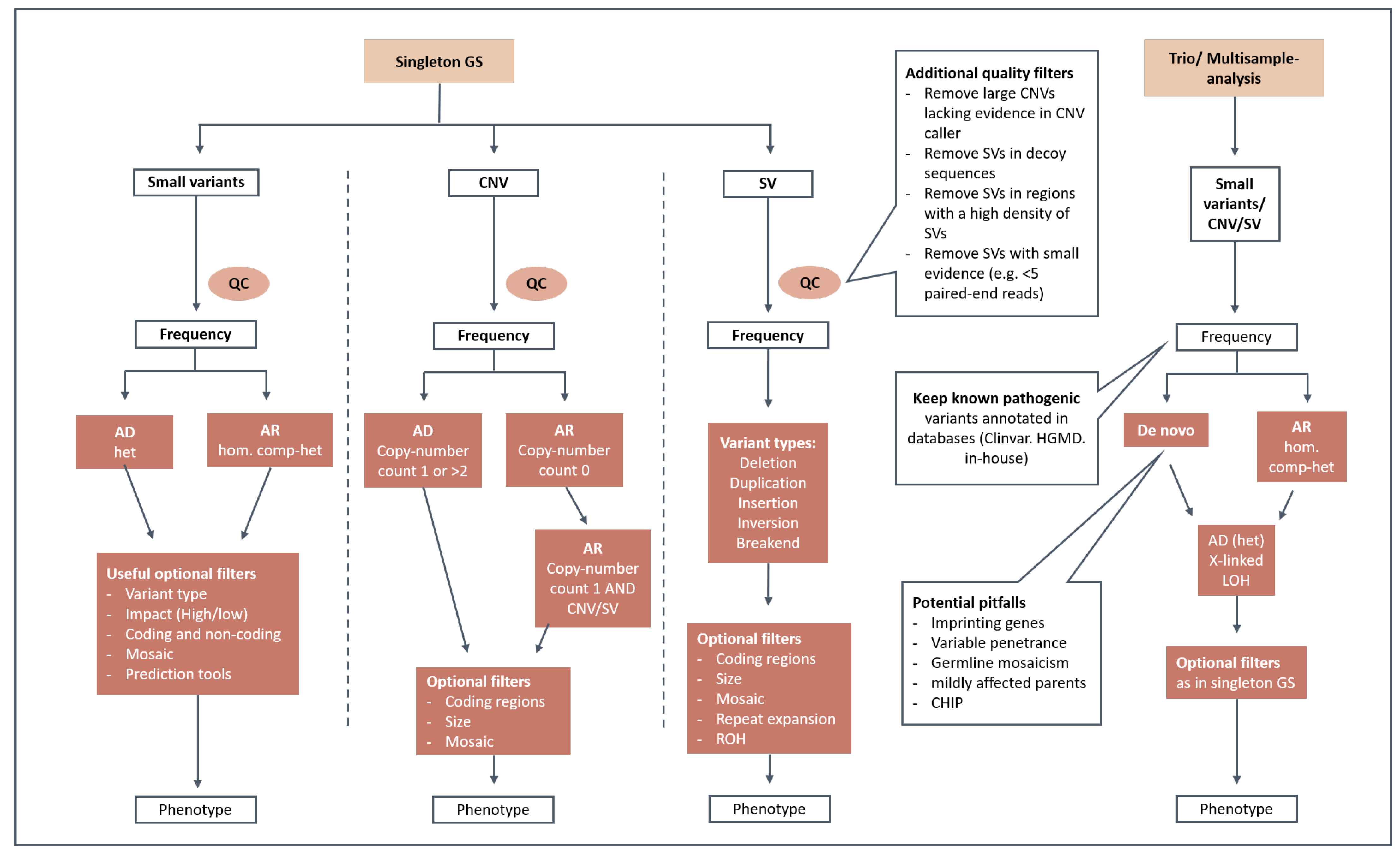
2.7. Diagnostic Filtering Strategies
2.8. Variant Interpretation
3. Results
3.1. Uniformity of Coverage and Gaps
3.2. Small Variant Calling Benchmarks
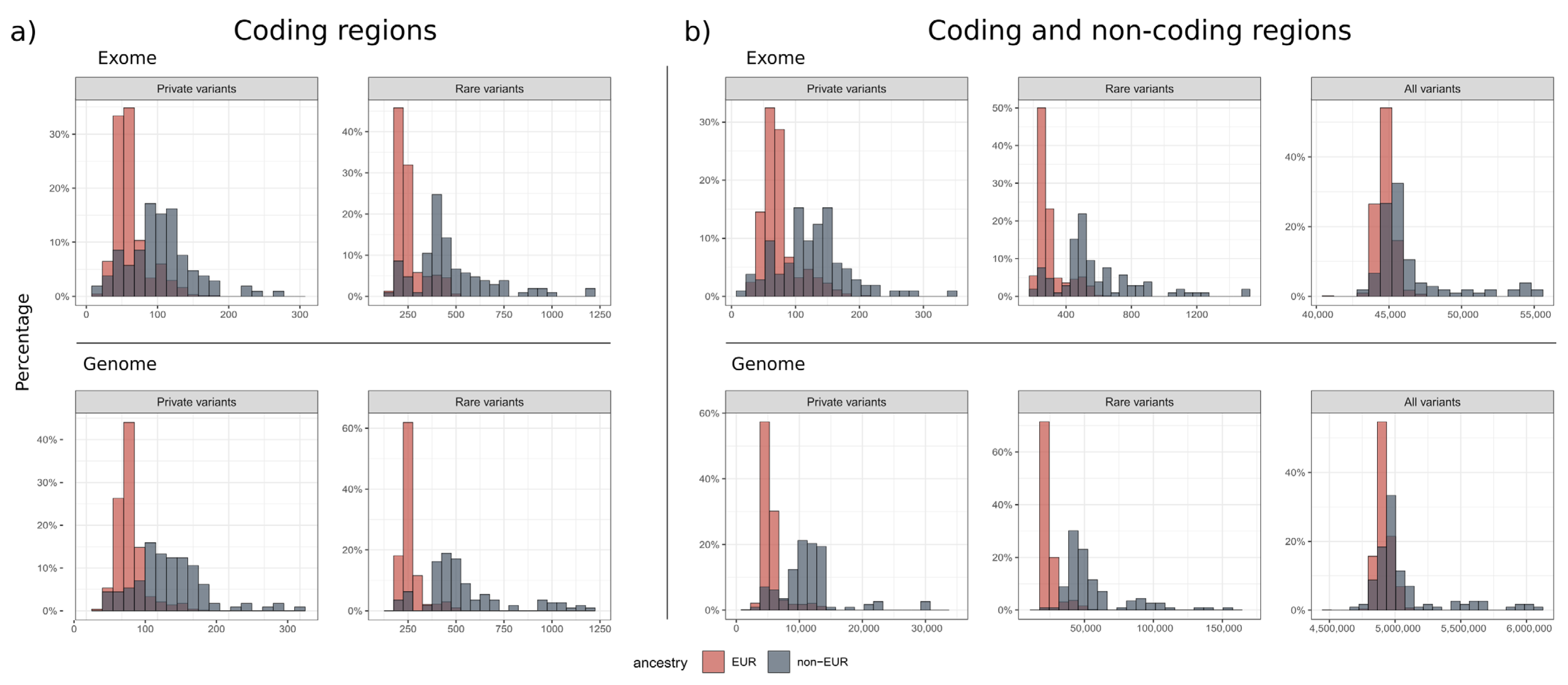
3.3. Number of Variants
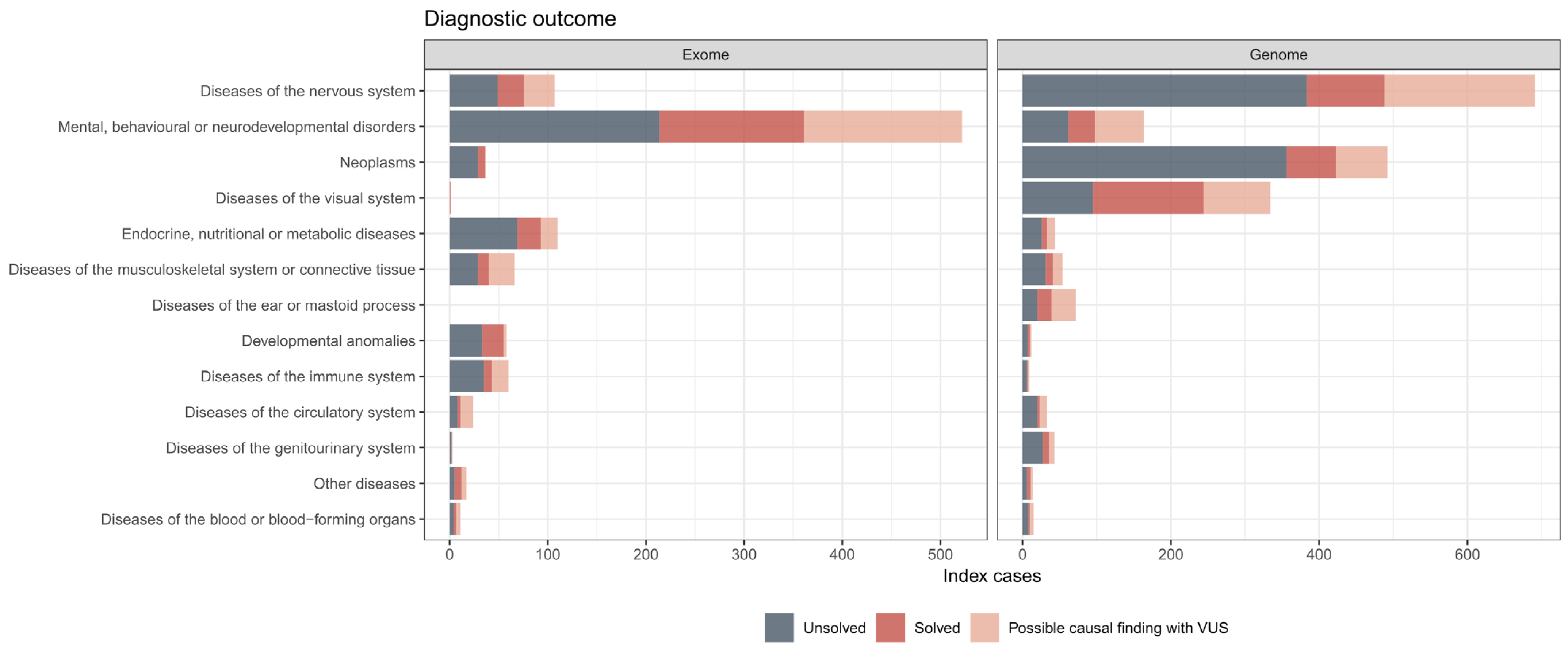
3.4. Diagnostic Outcomes in ES and GS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gilissen, C.; Hoischen, A.; Brunner, H.G.; Veltman, J.A. Unlocking Mendelian disease using exome sequencing. Genome Biol. 2011, 12, 228. [Google Scholar] [CrossRef] [PubMed]
- Gilissen, C.; Hehir-Kwa, J.Y.; Thung, D.T.; van de Vorst, M.; van Bon, B.W.M.; Willemsen, M.H.; Kwint, M.; Janssen, I.M.; Hoischen, A.; Schenck, A.; et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014, 511, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Riley, L.G.; Cowley, M.J.; Gayevskiy, V.; Minoche, A.E.; Puttick, C.; Thorburn, D.R.; Rius, R.; Compton, A.G.; Menezes, M.J.; Bhattacharya, K.; et al. The diagnostic utility of genome sequencing in a pediatric cohort with suspected mitochondrial disease. Genet. Med. 2020, 22, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Turro, E.; Astle, W.J.; Megy, K.; Gräf, S.; Greene, D.; Shamardina, O.; Allen, H.L.; Sanchis-Juan, A.; Frontini, M.; Thys, C.; et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature 2020, 583, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, T.; Scott, A.J.; Brandt, M.; Hall, I.M. Genomic Analysis in the Age of Human Genome Sequencing. Cell 2019, 177, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Marwaha, S.; Knowles, J.W.; Ashley, E.A. A guide for the diagnosis of rare and undiagnosed disease: Beyond the exome. Genome Med. 2022, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- van der Sanden, B.; Schobers, G.; Corominas Galbany, J.; Koolen, D.A.; Sinnema, M.; van Reeuwijk, J.; Stumpel, C.; Kleefstra, T.; de Vries, B.B.A.; Ruiterkamp-Versteeg, M.; et al. The performance of genome sequencing as a first-tier test for neurodevelopmental disorders. Eur. J. Hum. Genet. 2023, 31, 81–88. [Google Scholar] [CrossRef]
- Weisschuh, N.; Mazzola, P.; Zuleger, T.; Schaeferhoff, K.; Kuhlewein, L.; Kortum, F.; Witt, D.; Liebmann, A.; Falb, R.; Pohl, L.; et al. Diagnostic genome sequencing improves diagnostic yield: A prospective single-centre study in 1000 patients with inherited eye diseases. J. Med. Genet. 2023; ahead of print. [Google Scholar] [CrossRef]
- Wagner, J.; Olson, N.D.; Harris, L.; McDaniel, J.; Cheng, H.; Fungtammasan, A.; Hwang, Y.-C.; Gupta, R.; Wenger, A.M.; Rowell, W.J.; et al. Curated variation benchmarks for challenging medically relevant autosomal genes. Nat. Biotechnol. 2022, 40, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Dolzhenko, E.; Deshpande, V.; Schlesinger, F.; Krusche, P.; Petrovski, R.; Chen, S.; Emig-Agius, D.; Gross, A.; Narzisi, G.; Bowman, B.; et al. ExpansionHunter: A sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 2019, 35, 4754–4756. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD((R))): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Žemgulytė, A.; Applebaum, T.; Pritzel, A.; Wong, L.H.; Zielinski, M.; Sargeant, T.; et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 2023, 381, eadg7492. [Google Scholar] [CrossRef]
- Köhler, S.; Vasilevsky, N.A.; Engelstad, M.; Foster, E.; McMurry, J.; Aymé, S.; Baynam, G.; Bello, S.M.; Boerkoel, C.F.; Boycott, K.M.; et al. The Human Phenotype Ontology in 2017. Nucleic Acids Res. 2017, 45, D865–D876. [Google Scholar] [CrossRef]
- Corominas, J.; Smeekens, S.P.; Nelen, M.R.; Yntema, H.G.; Kamsteeg, E.; Pfundt, R.; Gilissen, C. Clinical exome sequencing-Mistakes and caveats. Hum. Mutat. 2022, 43, 1041–1055. [Google Scholar] [CrossRef]
- Brandt, T.; Sack, L.M.; Arjona, D.; Tan, D.; Mei, H.; Cui, H.; Gao, H.; Bean, L.J.H.; Ankala, A.; Del Gaudio, D.; et al. Adapting ACMG/AMP sequence variant classification guidelines for single-gene copy number variants. Genet. Med. 2020, 22, 336–344. [Google Scholar] [CrossRef]
- Richards, C.S.; Bale, S.; Bellissimo, D.B.; Das, S.; Grody, W.W.; Hegde, M.R.; Lyon, E.; Ward, B.E. ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet. Med. 2008, 10, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Aird, D.; Ross, M.G.; Chen, W.-S.; Danielsson, M.; Fennell, T.; Russ, C.; Jaffe, D.B.; Nusbaum, C.; Gnirke, A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011, 12, R18. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, S.; Singer-Berk, M.; Watts, N.A.; Phu, W.; Goodrich, J.K.; Solomonson, M.; Rehm, H.L.; MacArthur, D.G.; O’Donnell-Luria, A.; Genome Aggregation Database Consortium. Variant interpretation using population databases: Lessons from gnomAD. Hum. Mutat. 2022, 43, 1012–1030. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, H.; Korinth, D.; Ritthaler, M.; Schulte, B.; Battke, F.; von Kaisenberg, C.; Wüstemann, M.; Schulze, B.; Friedrich-Freksa, A.; Pfeiffer, L.; et al. Trio exome sequencing is highly relevant in prenatal diagnostics. Prenat. Diagn. 2022, 42, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Wortmann, S.B.; Oud, M.M.; Alders, M.; Coene, K.L.M.; van der Crabben, S.N.; Feichtinger, R.G.; Garanto, A.; Hoischen, A.; Langeveld, M.; Lefeber, D.; et al. How to proceed after “negative” exome: A review on genetic diagnostics, limitations, challenges, and emerging new multiomics techniques. J. Inherit. Metab. Dis. 2022, 45, 663–681. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.F.; Fitzgerald, T.W.; Jones, W.D.; Clayton, S.; McRae, J.F.; van Kogelenberg, M.; King, D.A.; Ambridge, K.; Barrett, D.M.; Bayzetinova, T.; et al. Genetic diagnosis of developmental disorders in the DDD study: A scalable analysis of genome-wide research data. Lancet 2015, 385, 1305–1314. [Google Scholar] [CrossRef]
- Incerti, D.; Xu, X.-M.; Chou, J.W.; Gonzaludo, N.; Belmont, J.W.; Schroeder, B.E. Cost-effectiveness of genome sequencing for diagnosing patients with undiagnosed rare genetic diseases. Genet. Med. 2022, 24, 109–118. [Google Scholar] [CrossRef]
- Jegathisawaran, J.; Tsiplova, K.; Hayeems, R.Z.; Marshall, C.R.; Stavropoulos, D.J.; Pereira, S.L.; Thiruvahindrapuram, B.; Liston, E.; Reuter, M.S.; Manshaei, R.; et al. Trio genome sequencing for developmental delay and pediatric heart conditions: A comparative microcost analysis. Genet. Med. 2022, 24, 1027–1036. [Google Scholar] [CrossRef]
- Wojcik, M.H.; Lemire, G.; Zaki, M.S.; Wissman, M.; Win, W.; White, S.; Weisburd, B.; Waddell, L.B.; Verboon, J.M.; VanNoy, G.E.; et al. Unique Capabilities of Genome Sequencing for Rare Disease Diagnosis. medRxiv 2023. [Google Scholar] [CrossRef]
- Yépez, V.A.; Gusic, M.; Kopajtich, R.; Mertes, C.; Smith, N.H.; Alston, C.L.; Ban, R.; Beblo, S.; Berutti, R.; Blessing, H.; et al. Clinical implementation of RNA sequencing for Mendelian disease diagnostics. Genome Med. 2022, 14, 38. [Google Scholar] [CrossRef] [PubMed]
- King, K.A.; Wegner, D.J.; Bucelli, R.C.; Shapiro, J.; Paul, A.J.; Dickson, P.I.; Wambach, J.A. Whole-Genome and Long-Read Sequencing Identify a Novel Mechanism in RFC1 Resulting in CANVAS Syndrome. Neurol. Genet. 2022, 8, e200036. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Tucci, A.; Cipriani, V.; Demidov, G.; Rocca, C.; Senderek, J.; Butryn, M.; Velic, A.; Lam, T.; Galanaki, E.; et al. Heterozygous UCHL1 loss-of-function variants cause a neurodegenerative disorder with spasticity, ataxia, neuropathy, and optic atrophy. Genet. Med. 2022, 24, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- Rafehi, H.; Read, J.; Szmulewicz, D.J.; Davies, K.C.; Snell, P.; Fearnley, L.G.; Scott, L.; Thomsen, M.; Gillies, G.; Pope, K.; et al. An intronic GAA repeat expansion in FGF14 causes the autosomal-dominant adult-onset ataxia SCA50/ATX-FGF14. Am. J. Hum. Genet. 2023, 110, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Greene, D.; Pirri, D.; Frudd, K.; Sackey, E.; Al-Owain, M.; Giese, A.P.J.; Ramzan, K.; Riaz, S.; Yamanaka, I.; Boeckx, N.; et al. Genetic association analysis of 77,539 genomes reveals rare disease etiologies. Nat. Med. 2023, 29, 679–688. [Google Scholar] [CrossRef]
- Wang, Q.; Dhindsa, R.S.; Carss, K.; Harper, A.R.; Nag, A.; Tachmazidou, I.; Vitsios, D.; Deevi, S.V.V.; Mackay, A.; Muthas, D.; et al. Rare variant contribution to human disease in 281,104 UK Biobank exomes. Nature 2021, 597, 527–532. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Exome | Genome | |
|---|---|---|
| Sample preparation | SureSelect Human All Exon v7 | TruSeq DNA PCR-Free kit |
| DNA-Sequencer system | NovaSeq 6000 system (Illumina) | NovaSeq 6000 system (Illumina) |
| Coverage aim | 150× | 38× |
| Coverage average | 157× | 39× |
| Coverage < 20× with MQ = 0 reads | 2.80% | 0.23% |
| Coverage < 20× with MQ ≥ 1 reads only | 3.26% | 0.63% |
| SNVs | InDels | ||||||
|---|---|---|---|---|---|---|---|
| F1 | Sensitivity | PPV | F1 | Sensitivity | PPV | ||
| GiaB high-confidence region, >15× coverage | ES | 0.9875 | 0.9906 | 0.9844 | 0.9427 | 0.9588 | 0.9273 |
| GS | 0.9955 | 0.9963 | 0.9947 | 0.9858 | 0.9778 | 0.9940 | |
| Protein-coding exons ± 20 bp, without coverage cutoff | ES | 0.9795 | 0.9720 | 0.9871 | 0.9309 | 0.9277 | 0.9341 |
| GS | 0.9916 | 0.9907 | 0.9924 | 0.9878 | 0.9869 | 0.9888 | |
| Exome | Genome | |||
|---|---|---|---|---|
| EUR | Non-EUR | EUR | Non-EUR | |
| Small variants (all) | 44,732 ± 595 | 46,504 ± 2778 | 4,913,537 ± 56,811 | 5,080,539 ± 292,286 |
| Small variants—rare (MAF ≤ 0.1%, NGSD count ≤ 10) | 302 ± 86 | 578 ± 253 | 24,809 ± 5655 | 58,732 ± 33,912 |
| Small variants—rare, coding (MAF ≤ 0.1%, NGSD count ≤ 10) | 252 ± 71 | 476 ± 203 | 260 ± 54 | 575 ± 318 |
| Small variants—private (MAF = 0%, NGSD count ≤ 1) | 73 ± 28 | 126 ± 56 | 5533 ± 1746 | 11,989 ± 7523 |
| Small variants—private, coding (MAF = 0%, NGSD count ≤ 1) | 61 ± 23 | 103 ± 46 | 61 ± 20 | 120 ± 69 |
| CNVs (all) | 10,727 ± 6520 | 13,668 ± 16,813 | ||
| CNVs—rare (AF ≤ 5%, good quality, OMIM) | 4 ± 6 | 16 ± 104 | ||
| SVs (all) | 10,907 ± 602 | 11,108 ± 792 | ||
| SVs—rare (AF ≤ 1%, good quality, OMIM) | 9 ± 4 | 21 ± 20 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.; Sturm, M.; Seibel-Kelemen, O.; Ossowski, S.; Haack, T.B. Lessons Learned from Translating Genome Sequencing to Clinical Routine: Understanding the Accuracy of a Diagnostic Pipeline. Genes 2024, 15, 136. https://doi.org/10.3390/genes15010136
Park J, Sturm M, Seibel-Kelemen O, Ossowski S, Haack TB. Lessons Learned from Translating Genome Sequencing to Clinical Routine: Understanding the Accuracy of a Diagnostic Pipeline. Genes. 2024; 15(1):136. https://doi.org/10.3390/genes15010136
Chicago/Turabian StylePark, Joohyun, Marc Sturm, Olga Seibel-Kelemen, Stephan Ossowski, and Tobias B. Haack. 2024. "Lessons Learned from Translating Genome Sequencing to Clinical Routine: Understanding the Accuracy of a Diagnostic Pipeline" Genes 15, no. 1: 136. https://doi.org/10.3390/genes15010136
APA StylePark, J., Sturm, M., Seibel-Kelemen, O., Ossowski, S., & Haack, T. B. (2024). Lessons Learned from Translating Genome Sequencing to Clinical Routine: Understanding the Accuracy of a Diagnostic Pipeline. Genes, 15(1), 136. https://doi.org/10.3390/genes15010136