
The Role of Next-Generation Sequencing in the Management of Patients with Suspected Non-Ischemic Cardiomyopathy after Syncope or Termination of Sudden Arrhythmic Death

Abstract
1. Introduction
2. Materials and Methods
Next-Generation Sequencing (NGS)
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peters, S.; Kumar, S.; Elliott, P.; Kalman, J.M.; Fatkin, D. Arrhythmic Genotypes in Familial Dilated Cardiomyopathy: Impli-cations for Genetic Testing and Clinical Management. Heart Lung Circ. 2019, 28, 31–38. [Google Scholar] [CrossRef]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. Group 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC). Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef] [PubMed]
- Punetha, J.; Hoffman, E.P. Short read (Next-Gen) sequencing: A tutorial with cardiomyopathy diagnostics as an exemplar. Circ. Cardiovasc. Genet. 2013, 6, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Fellmann, F.; van El, C.L.; Charron, P.; Michaud, P.; Howard, H.C.; Boers, S.N.; Clarke, A.J.; Duguet, A.; Forzano, F.; Kauferstein, S.; et al. European recommendations integrating genetic testing into multidisciplinary management of sudden cardiac death. Eur. J. Hum. Genet. 2019, 27, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Strande, N.T.; Riggs, E.R.; Buchanan, A.H.; Ceyhan-Birsoy, O.; DiStefano, M.; Dwight, S.S.; Goldstein, J.; Ghosh, R.; Seifert, B.A.; Sneddon, T.P.; et al. Evaluating the clinical validity of gene-disease asso-ciations: An evidence-based framework developed by the clinical genome resource. Am. J. Hum. Genet. 2017, 100, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Jordan, E.; Peterson, L.; Ai, T.; Asatryan, B.; Bronicki, L.; Brown, E.; Celeghin, R.; Edwards, M.; Fan, J.; Ingle, J.; et al. Evidence-Based Assessment of Genes in Dilated Cardiomyopathy. Circulation 2021, 144, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, L.; Bryant, R.M.; Vincent, G.M.; Flippin, M.; Lee, J.C.; Brown, E.; Zimmerman, F.; Rozich, R.; Szafranski, P.; et al. KCNQ1 mutations in patients with a family history of lethal cardiac arrhythmias and sudden death. Clin. Genet. 2003, 63, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Beavers, D.L.; Landstrom, A.P.; Chiang, D.Y.; Wehrens, X.H.T. Emerging roles of junctophilin-2 in the heart and implications for cardiac diseases. Cardiovasc. Res. 2014, 103, 198–205. [Google Scholar] [CrossRef]
- Haugaa, K.H.; Tilz, R.; Boveda, S.; Dobreanu, D.; Sciaraffia, E.; Mansourati, J.; Papiashvili, G.; Dagres, N. Implantable cardi-overter defibrillator use for primary prevention in ischaemic and non-ischaemic heart disease—Indications in the post-DANISH trial era. EP Eur. 2017, 19, 660–664. [Google Scholar]
- Poole, E.J. Present guidelines for device implantation: Clinical considerations and clinical challenges from pacing, implantable cardiac defibrillator, and cardiac resynchronization therapy. Circulation 2014, 129, 383–394. [Google Scholar] [CrossRef]
- Roberts, A.M.; Ware, J.S.; Herman, D.S.; Schafer, S.; Baksi, J.; Bick, A.G.; Buchan, R.J.; Walsh, R.; John, S.; Wilkinson, D.; et al. Integrated allelic, transcriptional, and phenomic dis-section of the cardiac effects of titin truncations in health and disease. Sci. Transl. Med. 2015, 7, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Zeppenfeld, K.; Tfelt-Hansen, J.; De Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; De Chillou, C.; et al. ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022, 43, 3997. [Google Scholar] [CrossRef] [PubMed]
- Broendberg, A.K.; Christiansen, M.K.; Nielsen, J.C.; Pedersen, L.N.; Jensen, H.K. Targeted next generation sequencing in a young population with suspected inherited malignant cardiac arrhythmias. Eur. J. Hum. Genet. 2018, 26, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.; Hamilton, R.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the chan-nelopathies and cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). EP Eur. 2011, 13, 1077–1109. [Google Scholar]
- Pottinger, T.D.; Puckelwartz, M.J.; Pesce, L.L.; Robinson, A.; Kearns, S.; Pacheco, J.A.; Rasmussen-Torvik, L.J.; Smith, M.E.; Chisholm, R.; McNally, E.M. Pathogenic and uncertain genetic variants have clinical correlates in diverse biobank participants. J. Am. Heart Assoc. 2020, 9, e013808. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Pua, C.J.; Bhalshankar, J.; Miao, K.; Walsh, R.; John, S.; Lim, S.Q.; Chow, K.; Buchan, R.; Soh, B.Y.; Lio, P.M.; et al. Development of a comprehensive sequencing assay for inherited cardiac condition genes. J. Cardiovasc. Transl. Res. 2016, 9, 3–11. [Google Scholar] [CrossRef]
- Mestroni, L.; Maisch, B.; McKenna, W.J.; Schwartz, K.; Charron, P.; Rocco, C.; Tesson, F.; Richter, A.; Wilke, A.; Komajda, M. Guidelines for the study of familial dilated cardiomyopathies: Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy. Eur. Heart J. 1999, 20, 93–102. [Google Scholar] [CrossRef]
- Ng, D.; Johnston, J.J.; Teer, J.K.; Singh, L.N.; Peller, L.C.; Wynter, J.S.; Lewis, K.L.; Cooper, D.N.; Stenson, P.D.; Mullikin, J.C.; et al. Interpreting secondary cardiac disease variants in an exome cohort. Circ. Cardiovasc. Genet. 2013, 6, 337–346. [Google Scholar] [CrossRef]
- Waldmüller, S.; Erdmann, J.; Binner, P.; Gelbrich, G.; Pankuweit, S.; Geier, C.; Timmermann, B.; Haremza, J.; Perrot, A.; Scheer, S.; et al. Novel correlations between the genotype and the phenotype of hypertrophic and dilated cardiomyopathy: Results from the German Competence Network Heart Failure. Eur. J. Heart Fail. 2011, 13, 1185–1192. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Y.; Zou, Y.; Sun, K.; Wang, Z.; Ding, H.; Yuan, J.; Wei, W.; Hou, Q.; Wang, H.; et al. Malignant effects of multiple rare variants in sarcomere genes on the prognosis of patients with hypertrophic cardiomyopathy. Eur. J. Heart Fail. 2014, 16, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Marcondes, L.; Crawford, J.; Earle, N.; Smith, W.; Hayes, I.; Morrow, P.; Donoghue, T.; Graham, A.; Love, D.; Skinner, J.R.; et al. Long QT molecular autopsy in sudden unexplained death in the young (1-40 years old): Lessons learnt from an eight year experience in New Zealand. PLoS ONE 2018, 13, e0196078. [Google Scholar] [CrossRef] [PubMed]
- Gladding, P.A.; Evans, C.A.; Crawford, J.; Chung, S.K.; Vaughan, A.; Webster, D.; Neas, K.; Love, D.R.; Rees, M.I.; Shelling, A.N.; et al. Posthumous diagnosis of long QT syndrome from neonatal screening cards. Heart Rhythm. 2010, 7, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, C.; Priori, S.G.; Schwartz, P.J.; Bloise, R.; Ronchetti, E.; Nastoli, J.; Bottelli, G.; Cerrone, M.; Leonardi, S. Genetic testing in the long QT syndrome: Development and validation of an efficient approach to genotyping in clinical practice. JAMA 2005, 294, 2975–2980. [Google Scholar] [CrossRef] [PubMed]
- Ware, J.S.; Walsh, R.; Cunningham, F.; Birney, E.; Cook, S.A. Paralogous annotation of disease-causing variants in long QT syndrome genes. Hum. Mutat. 2012, 33, 1188–1191. [Google Scholar] [CrossRef] [PubMed]
- Abunimer, A.; Smith, K.; Wu, T.J.; Lam, P.; Simonyan, V.; Mazumder, R. Single-Nucleotide Variations in Cardiac Arrhyth-mias: Prospects for Genomics and Proteomics Based Biomarker Discovery and Diagnostics. Genes 2014, 5, 254–269. [Google Scholar] [CrossRef]
- Burns, C.; Bagnall, R.D.; Lam, L.; Semsarian, C.; Ingles, J. Multiple Gene Variants in Hypertrophic Cardiomyopathy in the Era of Next-Generation Sequencing. Circ. Cardiovasc. Genet. 2017, 10, e001666. [Google Scholar] [CrossRef]
- Bollen, I.A.E.; van der Velden, J. The contribution of mutations in MYH7 to the onset of cardiomyopathy. Neth. Heart J. 2017, 25, 653–654. [Google Scholar] [CrossRef]
- Allegue, C.; Coll, M.; Mates, J.; Campuzano, O.; Iglesias, A.; Sobrino, B.; Brion, M.; Amigo, J.; Carracedo, A.; Brugada, P.; et al. Genetic Analysis of Arrhythmogenic Diseases in the Era of NGS: The Complexity of Clinical Deci-sion-Making in Brugada Syndrome. PLoS ONE 2015, 10, e0133037. [Google Scholar] [CrossRef]
- Aung, N.; Vargas, J.D.; Yang, C.; Cabrera, C.P.; Warren, H.R.; Fung, K.; Tzanis, E.; Barnes, M.R.; Rotter, J.I.; Taylor, K.D.; et al. Genome-Wide Analysis of Left Ventricular Image-Derived Phenotypes Identifies Fourteen Loci Associated with Cardiac Morphogenesis and Heart Failure Development. Circulation 2019, 140, 1318–1330. [Google Scholar] [CrossRef]
- Geier, C.; Gehmlich, K.; Ehler, E.; Hassfeld, S.; Perrot, A.; Hayess, K.; Cardim, N.; Wenzel, K.; Erdmann, B.; Krackhardt, F.; et al. Beyond the sarcomere: CSRP3 mutations cause hypertrophic cardiomyopathy. Hum. Mol. Genet. 2008, 17, 2753–2765. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.D.; Lakdawala, N.K.; Papoutsidakis, N.; Aubert, G.; Mazzanti, A.; McCanta, A.C.; Agarwal, P.P.; Arscott, P.P.; Dellefave-Castillo, L.M.; Vorovich, E.E.; et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct from Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020, 141, 1872–1884. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Behr, E.R.; Carrier, L.; van Duijn, C.; Evans, P.; Favalli, V.; van der Harst, P.; Haugaa, C.H.; Jondeau, G.; Kääb, S.; et al. Interpretation and actionability of genetic variants in cardiomyopathies: A position statement from the European Society of Cardiology Council on cardiovascular genomics. Eur. Heart J. 2022, 43, 1901–1916. [Google Scholar] [CrossRef] [PubMed]
- Sabater-Molina, M.; Navarro, M.; García-Molina Sáez, E.; Garrido, I.; Pascual-Figal, D.; González Carrillo, J.; Blanes, J.R.G. Mutation in JPH2 cause dilated cardiomyopathy. Clin. Genet. 2016, 90, 468–469. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, A.P.; Weisleder, N.; Batalden, K.B.; Bos, J.M.; Tester, D.J.; Ommen, S.R.; Wehrens, X.H.; Claycomb, W.C.; Ko, J.K.; Hwang, M.; et al. Mutations in JPH2-Encoded Junctophilin-2 Associated with Hypertrophic Car-diomyopathy in Humans. J. Mol. Cell Cardiol. 2007, 42, 1026–1035. [Google Scholar] [CrossRef]
- Lu, J.T.; Muchir, A.; Nagy, P.L.; Worman, H.J. LMNA cardiomyopathy: Cell biology and genetics meet clinical medicine. Dis. Model. Mech. 2011, 4, 562–568. [Google Scholar] [CrossRef]
- Warren, S.A.; Briggs, L.E.; Zeng, H.; Chuang, J.; Chang, E.I.; Terada, R.; Li, L.; Swanson, M.S.; Lecker, S.H.; Willis, M.S.; et al. Myosin Light Chain Phosphorylation Is Critical for Adaptation to Cardiac Stress. Circulation 2012, 126, 2575–2588. [Google Scholar] [CrossRef]
- Marshall, J.D.; Muller, J.; Collin, G.B.; Milan, G.; Kingsmore, S.F.; Dinwiddie, D.; Farrow, E.G.; Miller, M.A.; Favaretto, F.; Maffei, P.; et al. Alström Syndrome: Mutation spectrum of ALMS1. Hum. Mutat. 2015, 36, 660–668. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Shrivastava, S.; Ackerman, M.J.; Pereira, N.L. Clinical Impact of Secondary Risk Factors in TTN-Mediated Dilated Cardiomyopathy. Circ. Genom. Precis. Med. 2021, 14, e003240. [Google Scholar] [CrossRef]
- van Velzen, H.G.; Schinkel, A.F.L.; Oldenburg, R.A.; van Slegtenhorst, M.A.; Frohn-Mulder, I.M.E.; van der Velden, J.; Michels, M. Clinical Characteristics and Long-Term Outcome of Hypertrophic Cardiomyopathy in Individuals with a MYBPC3 (Myo-sin-Binding Protein C) Founder Mutation. Circ. Cardiovasc. Genet. 2017, 10, e001660. [Google Scholar] [CrossRef]
- Astrea, G.; Petrucci, P.; Cassandrini, D.; Savarese, M.; Trovato, R.; Lispi, L.; Rubegni, A.; Giacanelli, M.; Massa, R.; Nigro, V.; et al. Myoimaging in the NGS era: The discovery of a novel mutation in MYH7 in a family with distal myopathy and core-like features—A case report. BMC Med. Genet. 2016, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Fatkin, D.; Johnson, R. Variants of uncertaion Significance and «Misssing Patogeneicity». J. Am. Heart Assoc. 2020, 9, e015588. [Google Scholar] [CrossRef] [PubMed]
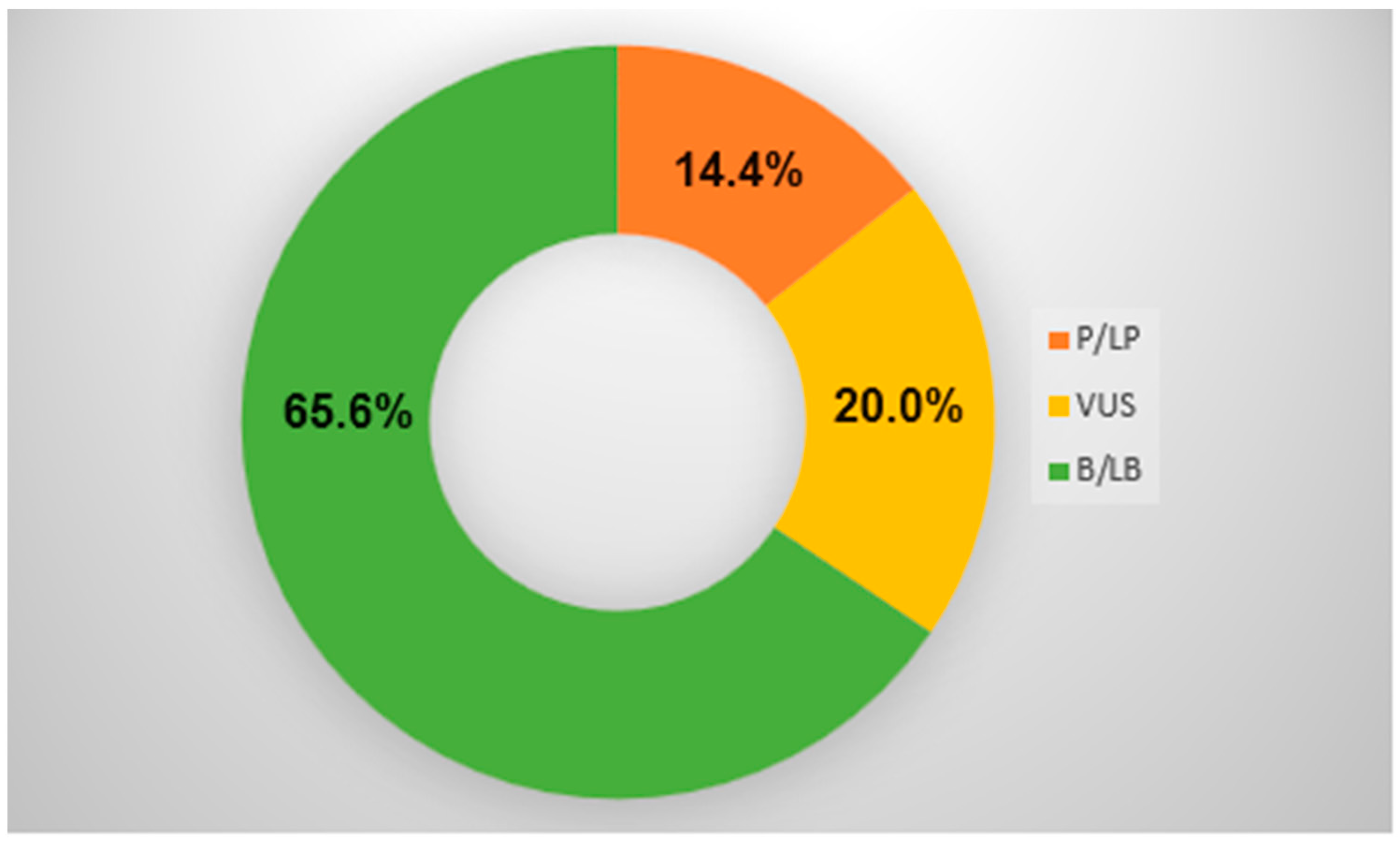

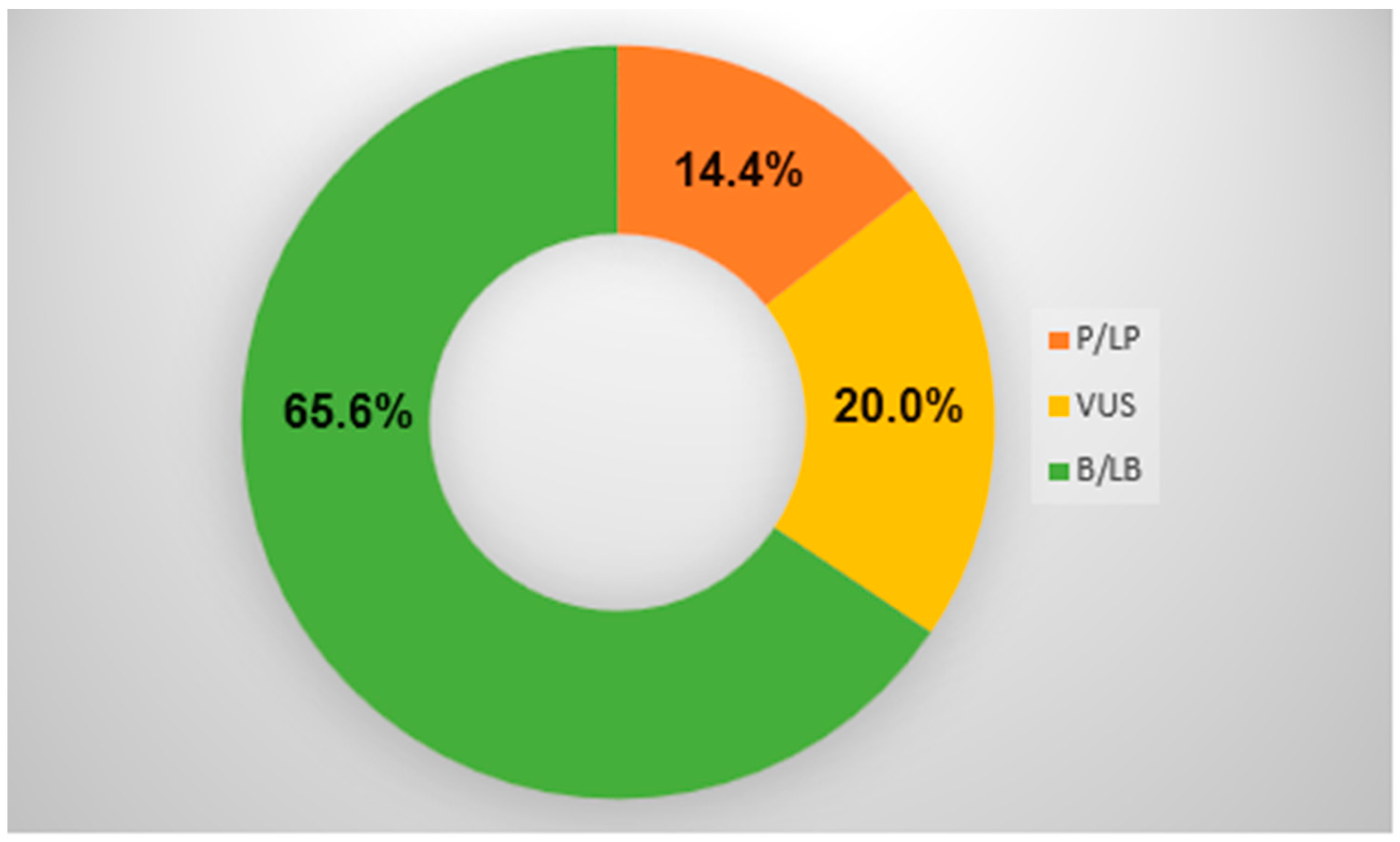{kind=link}
| Patient | Gene | Variant (Assembly UCSC, hg38) | Transcript Change | Protein Change | Variant Type |
|---|---|---|---|---|---|
| 1 | MYH7 | chr14:23429005 G-A | NM_000257.4:c.1357C>T | p.(Arg453Cys) | missense |
| 2 * | LMNA | chr1:g. 156136287 G-T | NM_170707.4:c.1231G>T | p.(Gly411Cys) | missense |
| 3 | MYBPC3 | chr11:g. 47335082 AG- | NM_000256.3:c.2864_2865delCT | p.(Pro955ArgfsTer95) | fremeshift |
| 4 * | LMNA | chr1:g. 156136287 G-T | NM_170707.4:c.1231G>T | p.(Gly411Cys) | missense |
| 5 * | JPH2 | chr20:g. 44115694 C- | NM_020433.4:c.1981delG | p.(Ala661ArgfsTer20) | fremeshift |
| 6 | MYBPC3 | chr11:g. 47337467 G-C | NM_000256.3:c.2526C>G | p.(Tyr842Ter) | sStop gain |
| 7 | SCN5A | chr3:g. 38606034 G-A | NM_198056.2:c.1255C>T | p.(Gln419Ter) | stop gain |
| 8 | KCNH2 | chr7:g. 150951679 C-T | NM_000238.3:c.1714G>A | p.(Gly572Ser) | missense |
| 9 | MYBPC3 | chr11:g. 47342718 C-T | NM_000256.3:c.1484G>A | p.(Arg495Gln) | missense |
| 10 | MYH6 | chr14:g. 23405122 C-A | NM_002471.3:c.508G>T | p.(Glu170Ter) | stop gain |
| 11 | TTN | chr2:g. 178575999 C-T | NM_001267550.1:c.70133G>A | p.(Trp23378Ter) | stop gain |
| 12 | VCL | chr10:g. 74071036 --T | NM_014000.3:c.452dup | p.(Glu152GlyfsTer19) | insertion |
| 13 * | MYBPC3 | chr11:g. 47343496 C-T | NM_000256.3:c.1219G>A | p.(Gly407Ser) | missense |
| 14 | LMNA | chr1:g. 156134819 C- | NM_170707.3:c.654delC | p.(Lys219SerfsTer261) | deletion |
| 15 | MYH7 | chr14:g. 23428516 A-G | NM_000257.2:c.1562T>C | p.(Ile521Thr) | missense |
| 16 | PKP2 | chr12:g. 32796145 C- | NM_004572.3:c.2453delG | p.(Gly818AlafsTer113) | deletion |
| 17 ** | KCNH2 | chr7:g. 150951530 G-C | NM_000238.4:c.1863C>G | p.(Ser621Arg) | missense |
| 18 | TTN | chr2:g. 178571794 G-A | NM_001256850.1:c.69415C>T | p.(Arg23139Ter) | stop gain |
| 19 | LMNA | chr1:g. 156134819 C- | NM_170707.3:c.654delC | p.(Lys219SerfsTer261) | deletion |
| 20 | DSP | chr6:g. 7555821 G-A | NM_004415.2:c.273+1G>A | p.? | alternative splicing |
| 21 | MYBPC3 | chr11:g. 47337467 G-C | NM_000256.3:c.2526C>G | p.(Tyr842Ter) | stop gain |
| 22 | GJA5 | chr1:g 147758631 T-G | NM_005266.6:c.608A>C | p.(Glu203Ala) | missense |
| 23 | LMNA | chr1:g 156134518 T-C | NM_170707.4:c.629T>C | p.(Ile210Thr) | missense |
| 24 * | RAF1 | chr3:g. 12585165 A-G | NM_002880.4:c.1625T>C | p.(Met542Thr) | missense |
| 25 | TTN | chr2:g. 178799908 C-A | NM_001267550.2:c.586G>T | p.(Glu196Ter) | stop gain |
| 26 | BRAF | chr7:g. 140801542 T-C | NM_004333.6:c.730A>G | p.(Thr244Ala) | missense |
| 27 | TTN | chr2:g. 178542496 G- | NM_001267550.2:c.97260del | p.(Trp32421GlyfsTer12) | fremeshift |
| 28 * | TTN | chr2:g. 178584435 A-G | NM_001267550.1:c.65116T>C | p.(Trp21706Arg) | missense |
| 29 | MYH7 | chr14:g. 23429262 A-T | NM_000257.2:c.1224T>A | p.(Asn408Lys) | missense |
| 30 * | MYBPC3 | chr11:g. 47335970 A-G | NM_000256.3:c.2644T>C | p.(Ser882Pro) | missense |
| Patient | Gene | Variant (Assembly UCSC, hg38) | Transcript Change | Protein Change | Variant Type |
|---|---|---|---|---|---|
| 1 | TTN | chr2:g. 178567143 C-T | NM_001267550.1:c.78989G>A | p.(Ser26330Asn) | missense |
| 2 | TTN | chr2:g. 178572357 C-G | NM_001267550.1:c.73775G>C | p.(Arg24592Thr) | missense |
| 3 | TTN | chr2:g. 178720616 C-A | NM_001267550.1:c.23146G>T | p.(Gly7716Cys) | missense |
| 4 | MYH6 | chr14:g. 23396970 G-A | NM_002471.3:c.2161C>T | p.(Arg721Trp) | missense |
| 5 | MYH6 | chr14:g. 23397017 C-T | NM_002471.3:c.2114G>A | p.(Arg705His) | missense |
| 6 | PRKAG2 | chr7:g. 151565804 T-C | NM_016203.3:c.1315A>G | p.(Ile439Val) | missense |
| 7 | KCNQ1 | chr11:g. 2588836 G-A | NM_000218.2:c.1375G>A | p.(Asp459Asn) | missense |
| 8 | LDB3 | chr10:g. 86718073 G-A | NM_001171610.1:c.1801G>A | p.(Val596Ile) | missense |
| 9 * | ACTA1 | chr1:g. 229431782 C-T | NM_001100.3:c.929G>A | p.(Gly310Glu) | missense |
| 10 | LDB3 | chr10:g. 86718073 G-A | NM_001171610.1:c.1801G>A | p.(Val601Ile) | missense |
| 11 | JUP | chr17:g. 41769629 C-T | NM_002230.4:c.257G>A | p.(Arg86Gln) | missense |
| 12 | TTN | chr2:g. 178567143 C-T | NM_001267550.1:c.78989G>A | p.(Ser26330Asn) | missense |
| 13 | KCND3 | chr1:g. 11177845 G-A | NM_004980.5:c.1501C>T | p.(Arg501Ter) | stop-gain |
| 14 | CASQ2 TTN | chr1:g. 115732965G-C chr2:g. 178545597 T-C | NM_001232.3:c.542C>G NM_001267550.1:c.95513A>G | p.(Ala181Gly) p.(Glu31838Gly) | missense missense |
| 15 | RYR2 | chr1:g. 237698992 G-C | NM_001035.3:c.9095G>C | p.(Cys3032Ser) | missense |
| 16 | KCNE3 | chr11:g. 74457269 G-A | NM_005472.4:c.295C>T | p.(Arg99Cys) | missense |
| 17 | RAF1 | chr3:g. 12611956 T-A | NM_002880.3:c.314A>T | p.(His105Leu) | missense |
| 18 | CRP3 | chr11:g. 19185024 G-A | NM_003476.5:c.436C>T | p.(Arg146Cys) | missense |
| 19 | TNNT2 MYH7 | chr1:g. 201363322T-C chr14:g. 23423938 A-G | NM_001276345.2:c.574A>G NM_000257.4:c.2891T>C | p.(Met192Val) p.(Val964Ala) | missense missense |
| 20 | MYH6 | chr14:g. 23389018 C-A | NM_002471.4:c.4016G>T | p.(Arg1339Leu) | missense |
| 21 | MYH11 | chr16:g. 15717232 C-T | NM_001040114.1:c.5433G>A | p.(Met1811Ile) | missense |
| 22 | DSP | chr6:g. 7579655 G-C | NM_004415.4:c.3465G>C | p.(Trp1155Cys) | missense |
| 23 | DSG2 | chr18:g. 31546084 G-C | NM_001943.5:c.2698G>C | p.(Glu900Gln) | missense |
| 24 | NEXN1 | chr1:g. 77942606 C-T | NM_144573.4:c.1805C>T | p.(Thr602Met) | missense |
| 25 | RYR2 | chr1:g. 237674110 C-T | NM_001035.3:c.8605C>T | p.(Pro2869Ser) | missense |
| 26 | CACNA1C | chr12:g. 2566445 G-A | NM_199460.3:c.1532G>A | p.(Arg511Gln) | missense |
| 27 | MYH6 | chr14:g. 23407028 C-T | NM_002471.4:c.196G>A | p.(Gly66Arg) | missense |
| 28 | TTN | chr2:g. 178718874 A-C | NM_001256850.1:c.23375T>G | p.(Val7792Gly) | missense |
| 29 | PKP2 | chr12:g. 32841047 T-C | NM_004572.4:c.1669A>G | p.(Asn557Asp) | missense |
| 30 | MYH11 | chr16:g. 15745209 T-C | NM_022844.2:c.2440A>G | p.(Thr814Ala) | missense |
| 31 | CACNA1C | chr12:g. 2053575 A-C | NM_199460.3:c.13A>C | p.(Asn5His) | missense |
| 32 | NOTCH1 | chr9:g. 136510685 T-C | NM_017617.5:c.2708A>G | p.(Asn903Ser) | missense |
| 33 | DSC2 | chr18:g.31074895 C-T | NM_024422.6:c.1676G>A | p.(Cys559Tyr) | missense |
| 34 | TNNI3 | chr19:g. 55157079 G-T | NM_000363.5:c.79C>A | p.(Arg27Ser) | missense |
| 35 ** | JUP | chr17:g. 41769150 G-A | NM_002230.4:c.526C>T | p.(Arg176Trp) | missense |
| 36 | MYH11 | chr16:g. 15784699 T-C | NM_001040114.1:c.653A>G | p.(Tyr218Cys) | missense |
| 37 | LMNA | chr1:g. 156135992 G-A | NM_170707.4:c.1028G>A | p.(Arg343Gln) | missense |
| 38 | LDB3 | chr10:g. 86692555 G-A | NM_001171610.2:c.1084G>A | p.(Ala362Thr) | missense |
| 39 ** | ACTC1 | chr15:g. 34792247 C-A | NM_005159.5:c.651C>T | p.(Lys217Asn) | missense |
| 40 | COL5A1 | chr9:g. 134700094 T-C | NM_001278074.1:c.463T>C | p.(Phe155Leu) | missense |
| 41 | DSP | chr6:g. 7585657 G-A | NM_004415.4:c.8395G>A | p.(Gly2799Arg) | missense |
| 42 | MYBPC3 | chr11:g. 47335970 A-G | NM_000256.3:c.2644T>C | p.(Ser882Pro) | missense |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vokač, D.; Stangler Herodež, Š.; Krgović, D.; Kokalj Vokač, N. The Role of Next-Generation Sequencing in the Management of Patients with Suspected Non-Ischemic Cardiomyopathy after Syncope or Termination of Sudden Arrhythmic Death. Genes 2024, 15, 72. https://doi.org/10.3390/genes15010072
Vokač D, Stangler Herodež Š, Krgović D, Kokalj Vokač N. The Role of Next-Generation Sequencing in the Management of Patients with Suspected Non-Ischemic Cardiomyopathy after Syncope or Termination of Sudden Arrhythmic Death. Genes. 2024; 15(1):72. https://doi.org/10.3390/genes15010072
Chicago/Turabian StyleVokač, Damijan, Špela Stangler Herodež, Danijela Krgović, and Nadja Kokalj Vokač. 2024. "The Role of Next-Generation Sequencing in the Management of Patients with Suspected Non-Ischemic Cardiomyopathy after Syncope or Termination of Sudden Arrhythmic Death" Genes 15, no. 1: 72. https://doi.org/10.3390/genes15010072
APA StyleVokač, D., Stangler Herodež, Š., Krgović, D., & Kokalj Vokač, N. (2024). The Role of Next-Generation Sequencing in the Management of Patients with Suspected Non-Ischemic Cardiomyopathy after Syncope or Termination of Sudden Arrhythmic Death. Genes, 15(1), 72. https://doi.org/10.3390/genes15010072