
Complete Genomic Sequence Analysis of a Sugarcane Streak Mosaic Virus Isolate from Yunnan Province of China


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material, Extraction and RT-PCR
2.2. Sequence Analysis
2.2.1. Sequence Assembly and Determination of Protease Cleavage Sites
2.2.2. Recombination Analysis, Phylogenetic and Genetic Distance Analysis
2.2.3. Phylogenetic and Genetic Distance Analysis
2.2.4. Selection Pressure Analysis
3. Results
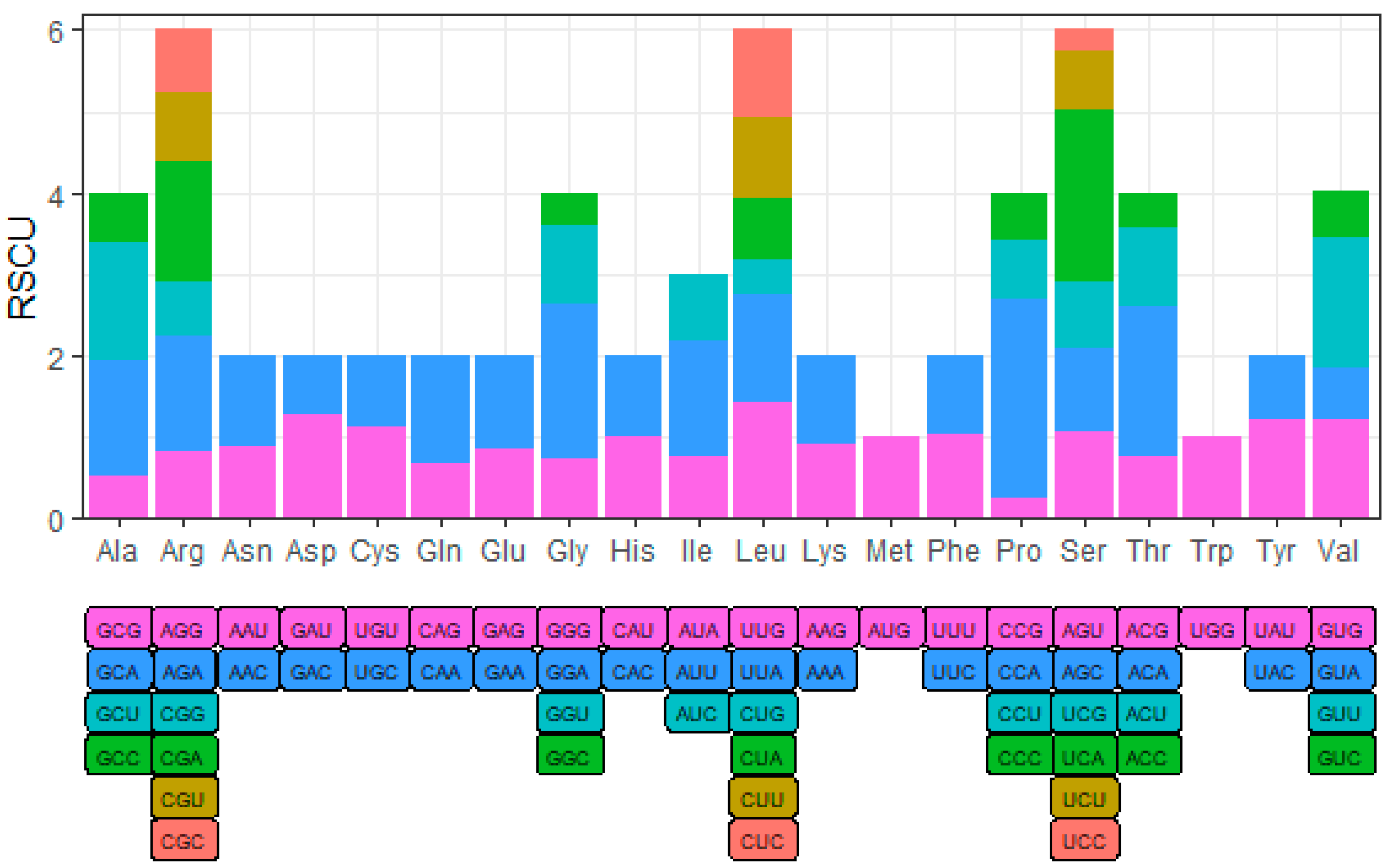
3.1. Genome Base Composition and Codon Preference of YN-21
3.2. YN-21 Protease Recognition Site Analysis
3.3. YN-21 Concordance Rate with 15 SCSMV Isolates
3.4. Recombination Analysis
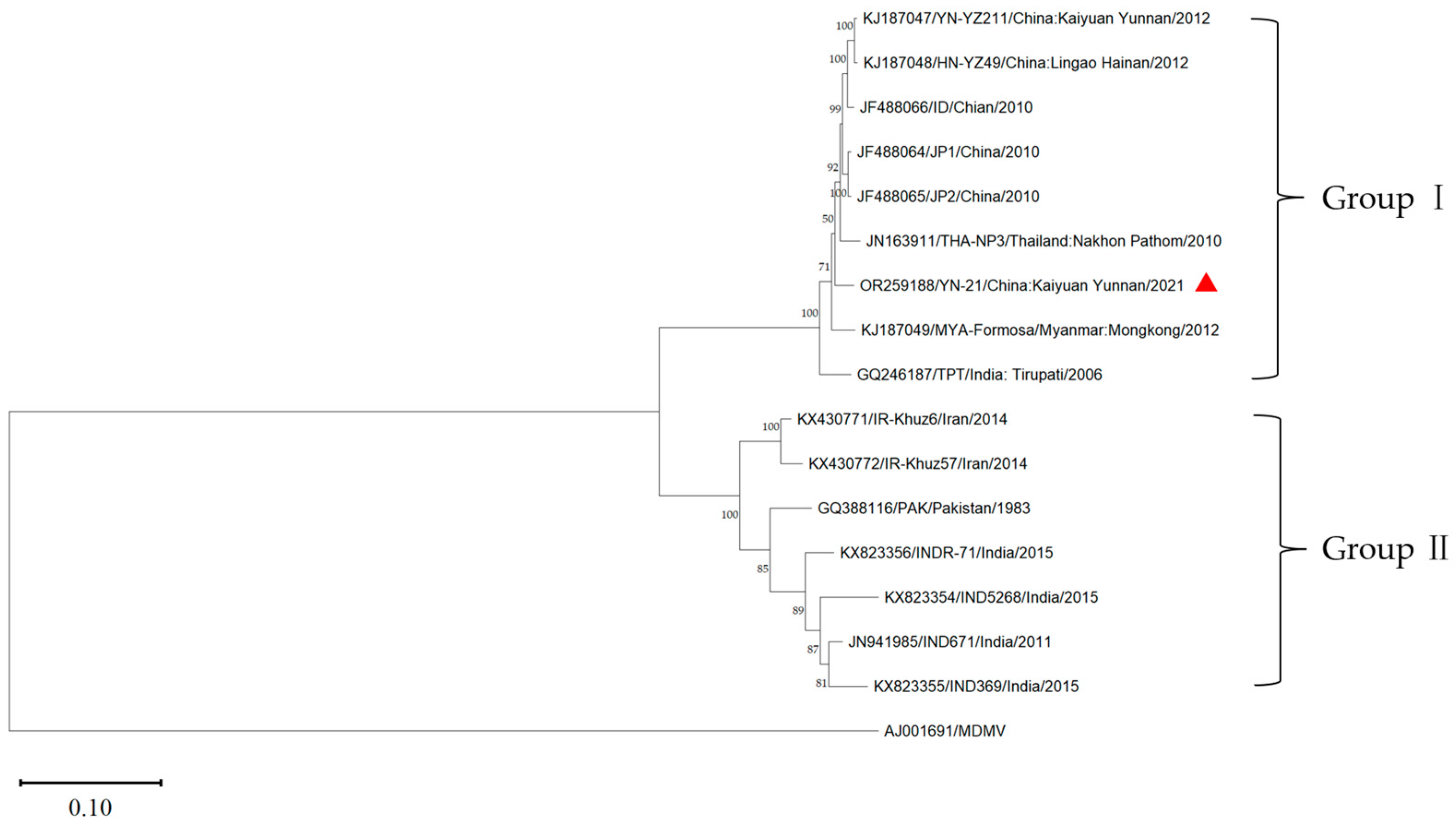
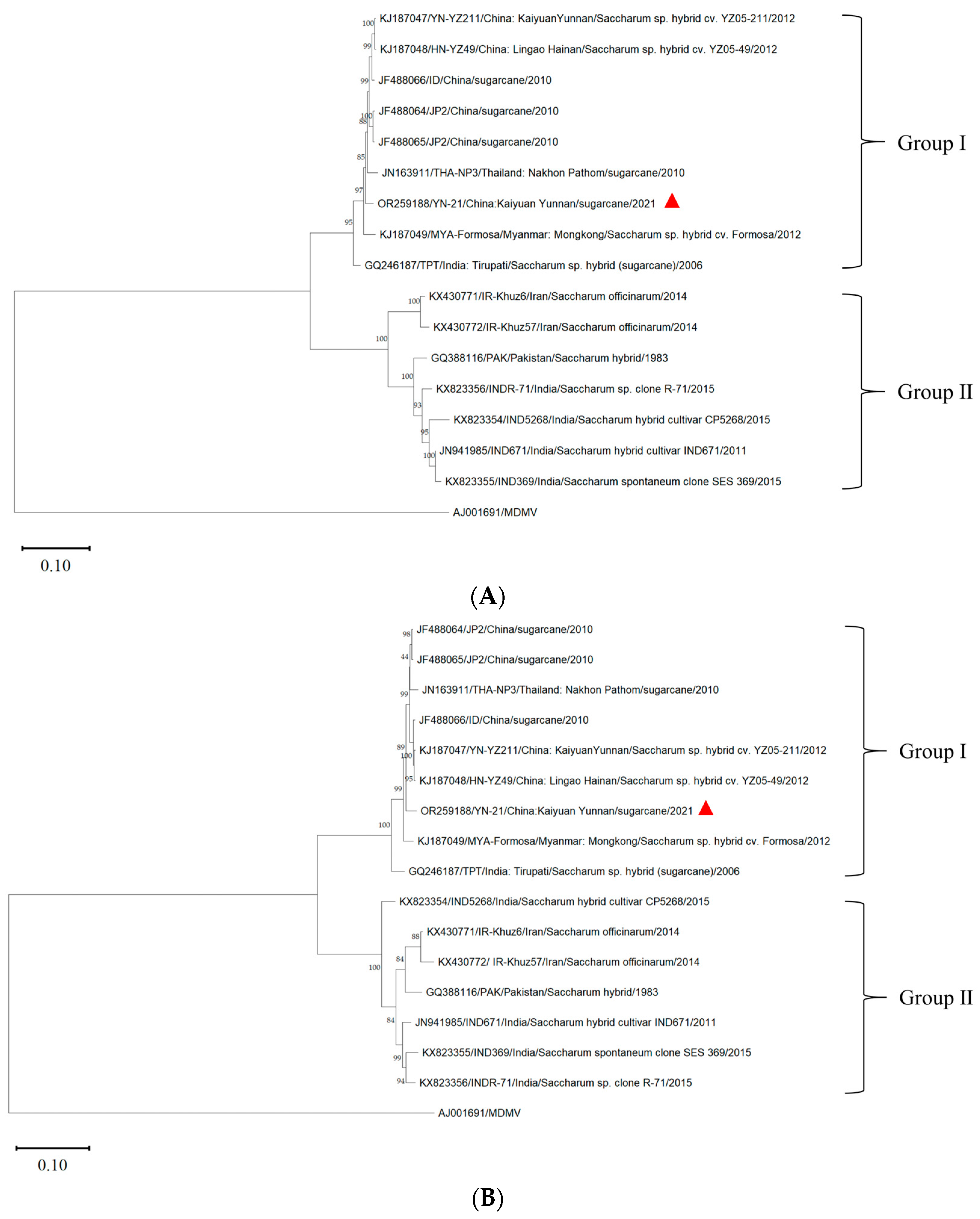
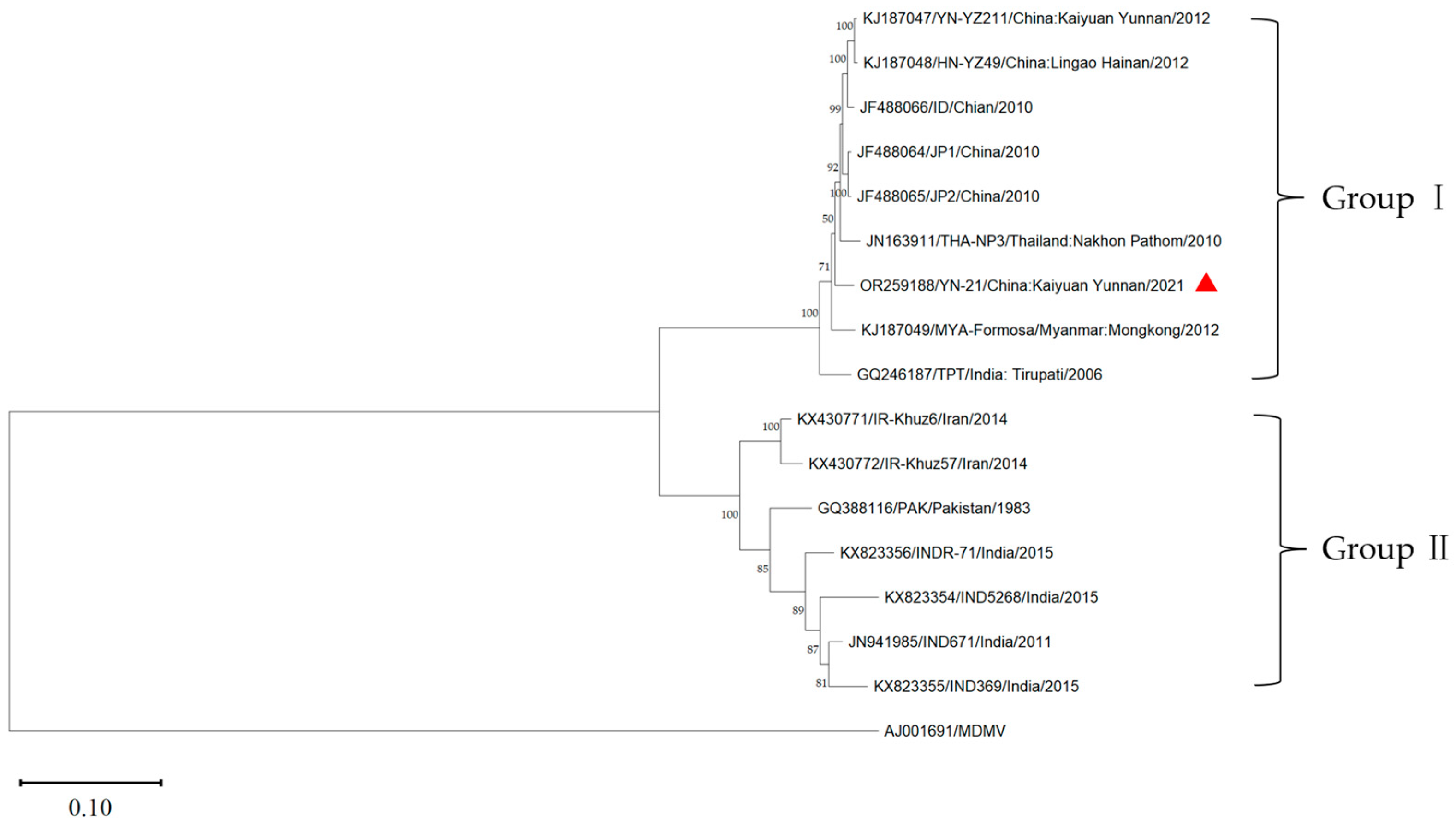
3.5. Phylogenetic Analysis
3.6. Selection Pressure Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

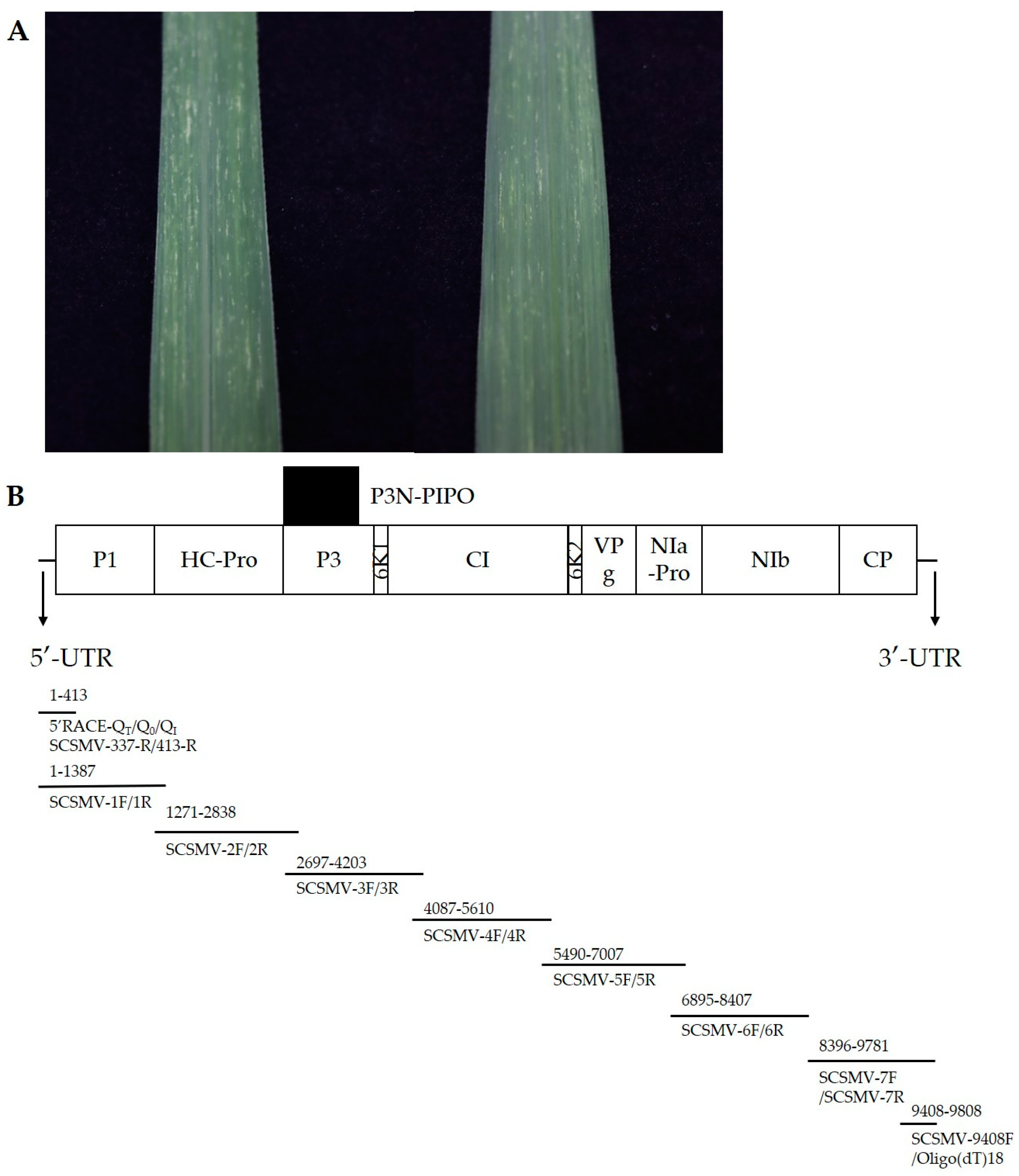{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Nucleotide Sequence (5′–3′) | Amplified Fragment /nt | Annealing Temperature/°C |
|---|---|---|---|
| SCSMV-1F | AAATGTAATTTCAAATTGACTACAATCA | 1–1387 | 55 |
| SCSMV-1R | CATGTTCTCGTAAGCTTCGTC | ||
| SCSMV-2F | TATACTGATGCTCATAAGGCAAG | 1271–2838 | 55 |
| SCSMV-2R | GACTCAGCTGTCCGATATTTG | ||
| SCSMV-3F | ACTTGCGAAGAATGATAGCATC | 2697–4203 | 55 |
| SCSMV-3R | CTAGCTGCGAGGTTAATCGAT | ||
| SCSMV-4F | TGTTATTGGTGGTGTTGGAAC | 4087–5610 | 55 |
| SCSMV-4R | GTGTTAAATAGGTTTGTCAACCG | ||
| SCSMV-5F | AAGTGGATAGAACTATTGGAATCTTAC | 5490–7007 | 55 |
| SCSMV-5R | AAGTGGATAGAACTATTGGAATCTTAC | ||
| SCSMV-6F | ATCATCATTCTACGTAAGTGAACAC | 6895–8407 | 55 |
| SCSMV-6R | CCTATGCGACATGTACTCGAC | ||
| SCSMV-7F | ATGTACCAAGATACGCGAATC | 8296–9781 | 55 |
| SCSMV-7R | CCTCCTCACTGGGCAGGTTG | ||
| SCSMV-9408F | CACCAGCAGAAATAGACGTGCGTAAC | 55 | |
| Oligo(dT)18 | CAGGATCCAAGCTTTTTTTTTTTTTTTTTT | ||
| 5′RACE-QT | CCAGTGAGCAGAGTGACGAGGACTCGAGCTCAAGCTTTTTTTTTTTTTTTTT | 55 | |
| 5′RACE-Q0 | CCAGTGAGCAGAGTGACG | ||
| 5′RACE-QI | GAGGACTCGAGCTCAAGC | ||
| SCSMV-413-R | CATATACTTCTGCTGCAATGTCG | 1–413 | |
| SCSMV-337-R | CTTACCTTTAACCGTCCAGAAGAC | 1–337 |
| Amino Acid | Abbreviation | Codon | Number | Amino Acid | Abbreviation | Codon | Number |
|---|---|---|---|---|---|---|---|
| Alanine | Ala | GCG | 27 | Proline | Pro | CCG | 9 |
| GCA | 74 | CCA | 83 | ||||
| GCU | 76 | CCU | 24 | ||||
| GCC | 31 | CCC | 20 | ||||
| Cystine | Cys | UGU | 29 | Glutarnine | Gln | CAG | 46 |
| UGC | 22 | CAA | 91 | ||||
| Aspartic acid | Asp | GAU | 112 | Arginine | Arg | AGG | 24 |
| GAC | 62 | AGA | 40 | ||||
| Glutamic acid | Glu | GAG | 84 | CGG | 19 | ||
| GAA | 112 | CGA | 42 | ||||
| Phenylalanine | Phe | UUU | 71 | CGU | 25 | ||
| UUC | 64 | CGC | 22 | ||||
| Glycine | Gly | GGG | 31 | Serine | Ser | AGU | 33 |
| GGA | 81 | AGC | 32 | ||||
| GGU | 41 | UCG | 26 | ||||
| GGC | 17 | UCA | 65 | ||||
| Histidine | His | CAU | 51 | UCU | 23 | ||
| CAC | 50 | UCC | 8 | ||||
| Isoleucine | Ile | AUA | 56 | Threonine | Thr | ACG | 42 |
| AUU | 103 | ACA | 101 | ||||
| AUC | 58 | ACU | 53 | ||||
| Lysine | Lys | AAG | 83 | ACC | 24 | ||
| AAA | 96 | Valine | Val | GUG | 61 | ||
| Leucine | Leu | UUG | 63 | GUA | 31 | ||
| UUA | 59 | GUU | 79 | ||||
| CUG | 18 | GUC | 26 | ||||
| CUA | 33 | Tryptophan | Trp | UGG | 49 | ||
| CUU | 44 | Tyrosine | Tyr | UAU | 70 | ||
| CUC | 47 | UAC | 44 | ||||
| Asparagine | Asn | AAU | 62 | Methionine | Met | AUG | 84 |
| AAC | 77 |
| Isolation | Complete Genome | CDS nt/aa | P1 | HC-Pro | P3 | 6K1 | CI | 6K2 | VPg | NIa-Pro | NIb | CP | P3N-PIPO |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| JF488066 | 97.43 | 97.66/99.30 | 98.32/99.72 | 97.02/98.51 | 98.28/99.70 | 98.64/100.00 | 97.61/99.70 | 97.92/97.92 | 96.30/98.99 | 97.34/99.58 | 97.94/99.20 | 97.75/98.93 | 98.18/98.91 |
| JF488065 | 97.42 | 97.65/99.30 | 98.32/99.72 | 97.02/98.30 | 97.98/99.39 | 99.32/100.00 | 97.36/99.54 | 97.92/100.00 | 96.97/98.99 | 97.34/99.58 | 97.94/99.40 | 97.98/99.29 | 98.42/98.91 |
| JF488064 | 97.41 | 97.64/99.30 | 98.23/99.72 | 97.02/98.3 | 97.88/99.39 | 99.32/100.00 | 97.41/99.70 | 97.92/100.00 | 96.80/98.99 | 97.34/99.58 | 98.07/99.40 | 97.86/98.93 | 98.30/98.91 |
| KJ187047 | 97.08 | 97.46/99.23 | 98.23/99.72 | 97.09/98.72 | 98.28/99.39 | 97.28/100.00 | 96.75/99.39 | 96.53/100.00 | 96.30/98.48 | 97.20/99.16 | 98.01/99.40 | 97.98/98.93 | 98.18/98.91 |
| KJ187048 | 97.04 | 97.40/99.30 | 97.86/99.44 | 96.81/98.30 | 98.08/99.70 | 97.96/100.00 | 96.85/99.70 | 96.53/100.00 | 96.63/98.99 | 97.20/99.58 | 97.94/94.82 | 97.75/98.93 | 98.30/98.91 |
| JN163911 | 96.85 | 97.01/99.17 | 96.46/98.88 | 96.74/98.51 | 97.37/99.09 | 98.64/100.00 | 96.80/99.54 | 98.61/100.00 | 95.79/98.48 | 96.92/99.58 | 97.14/99.60 | 98.34/98.93 | 97.69/98.91 |
| KJ187049 | 96.57 | 96.86/99.20 | 96.55/99.16 | 96.60/98.51 | 97.07/98.79 | 96.60//97.96 | 96.44/99.70 | 97.22/100.00 | 96.46/98.48 | 96.92/99.16 | 97.01/99.80 | 97.98/98.58 | 97.57/98.91 |
| GQ246187 | 94.83 | 96.22/98.75 | 96.37/99.16 | 96.31/98.09 | 97.47/98.79 | 80.27/100.00 | 97.36/99.39 | 97.92/100.00 | 95.62/98.48 | 90.90/97.48 | 96.68/99.20 | 94.42/97.86 | 98.42/98.91 |
| KX430772 | 82.13 | 81.82/94.25 | 83.61/94.13 | 76.60/87.45 | 85.45/94.55 | 76.87/93.88 | 81.00/95.88 | 75.69/100.00 | 82.66/97.47 | 83.05/97.06 | 81.47/94.22 | 86.71/96.09 | 87.23/92.34 |
| KX823354 | 82.01 | 81.72/92.59 | 79.87/83.38 | 76.45/86.26 | 84.65/94.55 | 80.27/91.84 | 81.71/95.58 | 75.69/100.00 | 81.48/97.98 | 82.52/94.12 | 81.54/94.42 | 95.10/95.73 | 86.50/91.97 |
| KX823355 | 81.55 | 81.34/92.12 | 80.33/85.87 | 77.52/89.57 | 83.43/91.52 | 77.55/85.71 | 80.67/93.46 | 76.39/100.00 | 81.82/96.46 | 81.09/91.67 | 80.35/93.06 | 91.34/94.66 | 82.27/84.75 |
| KX430771 | 81.67 | 81.34/94.82 | 83.71/95.25 | 76.60/89.57 | 85.05/94.24 | 74.83/91.84 | 80.54/96.80 | 74.31/100.00 | 81.99/97.98 | 83.05/97.48 | 80.35/94.62 | 85.88/94.66 | 86.74/92.34 |
| GQ388116 | 81.60 | 81.25/95.11 | 82.31/94.97 | 77.80/90.00 | 84.95/94.85 | 80.27/93.88 | 79.67/97.41 | 75.69/100.00 | 81.14/96.97 | 82.63/97.06 | 80.61/95.02 | 86.12/95.37 | 86.25/91.24 |
| KX823356 | 81.48 | 81.18/93.58 | 81.90/88.37 | 76.95/88.79 | 84.34/93.64 | 80.95/91.84 | 78.81/96.49 | 76.39/100.00 | 81.99/97.47 | 80.39/92.86 | 80.81/94.82 | 97.50/95.37 | 86.01/90.51 |
| JN941985 | 81.32 | 80.98/87.81 | 81.53/87.81 | 77.52/90.21 | 84.75/95.76 | 80.95/91.84 | 79.78/97.41 | 75.69/100.00 | 80.98/97.98 | 81.37/94.54 | 80.41/94.82 | 86.12/96.09 | 86.01/90.88 |
References
- Xu, D.L.; Zhou, G.H.; Xie, Y.J.; Mock, R.; Li, R. Complete Nucleotide Sequence and Taxonomy of Sugarcane Streak Mosaic Vi-rus, Member of a Novel Genus in the Family Potyviridae. Virus Genes 2010, 40, 432–439. [Google Scholar] [CrossRef]
- Xu, D.L.; Park, J.W.; Mirkov, T.E.; Zhou, G.H. Viruses Causing Mosaic Disease in Sugarcane and Their Genetic Diversity in Southern China. Arch. Virol. 2008, 153, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Putra, L.K.; Kristini, A.; Achadian, E.M.; Damayanti, T. Sugarcane Streak Mosaic Virus in Indonesia: Distribution, Characterisation, Yield Losses and Management Approaches. Sugar Tech. 2014, 16, 392–399. [Google Scholar] [CrossRef]
- He, E.Q.; Bao, W.Q.; Sun, S.R.; Hu, C.Y.; Chen, J.S.; Bi, Z.W.; Xie, Y.; Lu, J.J.; Gao, S.J. Incidence and Distribution of Four Viruses Causing Diverse Mosaic Diseases of Sugarcane in China. Agronomy 2022, 12, 302. [Google Scholar] [CrossRef]
- Singh, D.; Rao, G.P. Sudan Grass (Sorghum sudanense Stapf): A New Sugarcane Streak Mosaic Virus Mechanical Host. Guangxi Agric. Sci. 2010, 41, 436–438. [Google Scholar]
- He, Z.; Yasaka, R.; Li, W.; Li, S.; Ohshima, K. Genetic Structure of Populations of Sugarcane Streak Mosaic Virus in China: Comparison with the Populations in India. Virus Res. 2016, 211, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.S.; Alabi, O.J.; Damaj, M.B.; Fu, W.L.; Sun, S.R.; FU, H.Y.; Chen, R.K.; Mirkov, T.E.; Gao, S.J. Genomic Variability and Molecular Evolution of Asian Isolates of Sugarcane Streak Mosaic Virus. Arch. Virol. 2016, 161, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Parameswari, B.; Bagyalakshmi, K.; Chinnaraja, C.; Viswanathan, R. Molecular Characterization of Indian Sugarcane Streak Mosaic Virus Isolates Reveals Recombination and Negative Selection in the P1 Gene. Gene 2014, 552, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Moradi, Z.; Mehrvar, M.; Nazifi, E. Genetic Diversity and Biological Characterization of Sugarcane Streak Mosaic Virus Isolates from Iran. Virusdisease 2018, 29, 316–323. [Google Scholar] [CrossRef]
- Sorho, F.; Sereme, D.; Kouamé, K.D.; Koné, N.; Yao, K.J.E.; Ouattara, M.M.; Tapsoba, W.P.; Ouattara, B.; Kone, D. First Report of Sugarcane Streak Mosaic Virus (SCSMV) Infecting Sugarcane in Côte d’Ivoire. Plant Dis. 2020, 105, 519. [Google Scholar] [CrossRef]
- Lu, G.; Wang, Z.; Xu, F.; Pan, Y.B.; Grisham, M.P.; Xu, L. Sugarcane Mosaic Disease: Characteristics, Identification and Control. Microorganisms 2021, 9, 1984. [Google Scholar] [CrossRef]
- Hema, M.; Joseph, J.; Gopinath, K.; Sreenivasulu, P.; Savithri, H.S. Molecular Characterization and Interviral Relationships of a Flexuous Filamentous Virus Causing Mosaic Disease of Sugarcane (Saccharum officinarum L.) in India. Arch. Virol. 1999, 144, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Urcuqui-Inchima, S.; Haenni, A.L.; Bernardi, F. Potyvirus Proteins: A Wealth of Functions. Virus Res. 2001, 74, 157–175. [Google Scholar] [CrossRef] [PubMed]
- Riechmann, J.L.; Lain, S.; Garcia, J.A. Highlights and Prospects of Potyvirus Molecular Biology. J. Gen. Virol. 1992, 73, 1–16. [Google Scholar] [CrossRef]
- Chung, B.Y.; Miller, W.A.; Atkins, J.F.; Firth, A.E. An Overlapping Essential Gene in the Potyviridae. Proc. Natl. Acad. Sci. USA 2008, 105, 5897–5902. [Google Scholar] [CrossRef]
- Ogawa, T.; Tomitaka, Y.; Nakagawa, A.; Ohshima, K. Genetic Structure of a Population of Potato Virus Y Inducing Potato Tuber Necrotic Ringspot Disease in Japan; Comparison with North American and European Populations. Virus Res. 2008, 131, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Gillaspie, A.G., Jr.; Mock, R.G.; Smith, F.F. Identification of sugarcane mosaic virus and characterization of strains of the virus from Pakistan, Iran, and Camaroon. Proc. Int. Soc. Sugar Cane Technol. 1978, 16, 347–355. [Google Scholar]
- Hema, M.; Savithri, H.S.; Sreenivasulu, P.; Rao, G.P.; Ford, R.E.; Tošić, M.; Teakle, D.S. Sugarcane Streak Mosaic virus: Occurrence, Purification, Characterization and Detection. In Sugarcane Pathology; Science Publishers, Inc.: Enfield, CT, USA, 2001. [Google Scholar]
- Wang, X.Y.; Li, W.F.; Huang, Y.K.; Zhang, R.Y.; Shan, H.L.; Yin, J.; Luo, Z.M. Molecular Detection and Phylogenetic Analysis of Viruses Causing Mosaic Symptoms in New Sugarcane Varieties in China. Eur. J. Plant Pathol. 2017, 148, 931–940. [Google Scholar] [CrossRef]
- Zhang, R.Y.; Li, W.F.; Huang, Y.K.; Pu, C.H.; Wang, X.Y.; Shan, H.L.; Cang, X.Y.; Luo, Z.M.; Yin, J. Genetic Diversity and Population Structure of Sugarcane Streak Mosaic Virus in Yunnan Province, China. Trop. Plant Pathol. 2018, 43, 514–519. [Google Scholar] [CrossRef]
- Bagyalakshmi, K.; Parameswari, B.; Chinnaraja, C.; Karuppaiah, R.; Ganesh Kumar, V.; Viswanathan, R. Genetic Variability and Potential Recombination Events in the HC-Pro Gene of Sugarcane Streak Mosaic Virus. Arch. Virol. 2012, 157, 1371–1375. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Adams, M.J.; Antoniw, J.F.; Beaudoin, F. Overview and Analysis of the Polyprotein Cleavage Sites in the Family Potyviridae. Mol. Plant Pathol. 2005, 6, 471–487. [Google Scholar] [CrossRef]
- Wei, T.Y.; Zhang, C.W.; Hong, J.; Xiong, R.Y.; Kasschau, K.D.; Zhou, X.P.; Carrington, J.C.; Wang, A.M. Formation of Complexes at Plasmodesmata for Potyvirus Intercellular Movement is Mediated by the Viral Protein P3N-PIPO. PLoS Pathog. 2010, 6, e1000962. [Google Scholar] [CrossRef]
- Martin, D.P.; Rybicki, E. RDP: Detection of Recombination Amongst Aligned Sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Sawyer, S. Statistical tests for detecting gene conversion. Mol. Biol. Evol. 1989, 6, 526–538. [Google Scholar] [PubMed]
- Salminen, M.O.; Carr, J.K.; Burke, D.S.; McCutchan, F.E. Identification of Breakpoints in Intergenotypic Recombinants of HIV Type 1 by Bootscanning. AIDS Res. Hum. Retroviruses 1995, 11, 1423–1425. [Google Scholar] [CrossRef]
- Smith, J.M. Analyzing the Mosaic Structure of Genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. Evaluation of Methods for Detecting Recombination from DNA Sequences: Computer Simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo Procedure for Assessing Signals in Recombinant Sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Lemey, P.; Posada, D. Analysing Recombination in Nucleotide Sequences. Mol. Ecol. Resour. 2011, 11, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Felsenstein, J. Evolutionary trees from DNA sequences: A maximum likelihood approach. J. Mol. Evol. 1981, 17, 368–376. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so Different after all: A Comparison of Methods for Detecting Amino Acid Sites under Selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Stadler, M.; Fire, A. Wobble base-pairing slows in vivo translation elongation in metazoans. RNA 2011, 17, 2063–2073. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Stapleton, S.C.; Yang, M.T.; Cha, S.S.; Choi, C.K.; Galie, P.A.; Chen, C.S. Biomimetic Model to Reconstitute Angiogenic Sprouting Morphogenesis in Vitro. Proc. Natl. Acad. Sci. USA 2013, 110, 6712–6717. [Google Scholar] [CrossRef]
- Singhal, P.; Nabi, S.U.; Yadav, M.K.; Dubey, A. Mixed Infection of Plant Viruses: Diagnostics, Interactions and Impact on Host. J. Plant Dis. Prot. 2021, 128, 353–368. [Google Scholar] [CrossRef]
- Delsuc, F.; Brinkmann, H.; Philippe, H. Phylogenomics and the Reconstruction of the Tree of Life. Nat. Rev. Genet. 2005, 6, 361–375. [Google Scholar] [CrossRef]
- Viswanathan, R.; Balamuralikrishnan, M.; Karuppaiah, R. Characterization and Genetic Diversity of Sugarcane Streak Mosaic Virus Causing Mosaic in Sugarcane. Virus Genes. 2008, 36, 553–564. [Google Scholar] [CrossRef]
- Li, W.F.; He, Z.; Li, S.F.; Huang, Y.K.; Zhang, Z.X.; Jiang, D.M.; Wang, X.Y.; Luo, Z.M. Molecular Characterization of a New Strain of Sugarcane Streak Mosaic Virus (SCSMV). Arch. Virol. 2011, 156, 2101–2104. [Google Scholar] [CrossRef]
- Harborne, K.M. Population Dynamics of the Main Aphid Vectors of Sugarcane Mosaic Virus in Natal; International Society of Sugar Cane Technologists: Sao Paulo, Brazil, 1988. [Google Scholar]
- Aung, N.N.; Khaing, E.E.; Mon, Y.Y. History of Sugarcane Breeding, Germplasm Development and Related Research in Myanmar. Sugar Tech. 2022, 24, 243–253. [Google Scholar] [CrossRef]
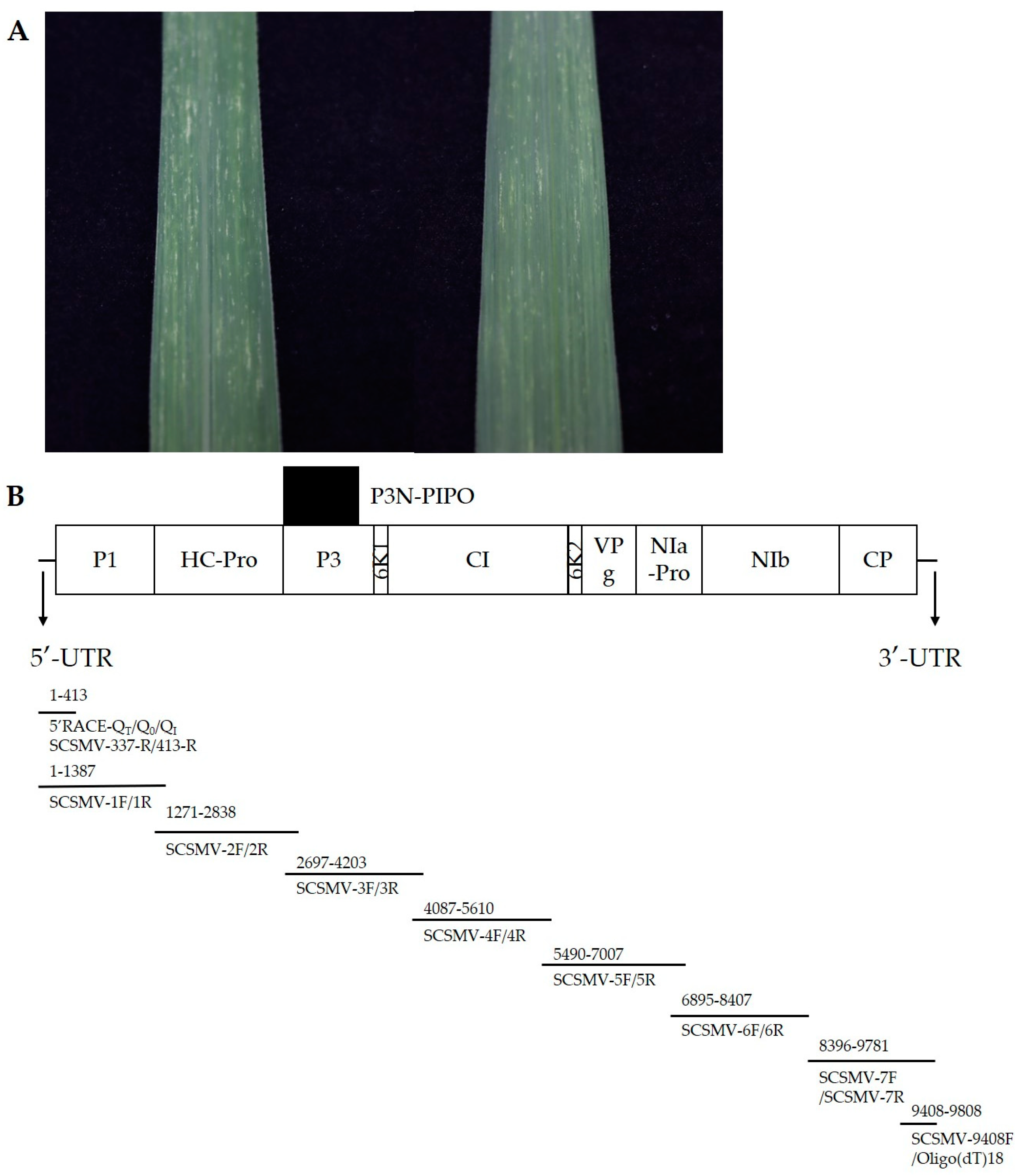
| Genome Fragment | Start-End Site | Size in nt/aa | Cleavage Site (C-Terminus) |
|---|---|---|---|
| 5′-UTR | 1–199 | 199 | |
| P1 | 200–1273 | 1074/358 | EDLVFY/T |
| HC-Pro | 1274–2683 | 1410/470 | MKYRIG/G |
| P3 | 2684–3673 | 990/330 | LVHHAQ/G |
| 6K1 | 3674–3820 | 147/49 | TTSSPE/S |
| CI | 3821–5788 | 1968/656 | IYHGGQ/E |
| 6K2 | 5789–5932 | 144/48 | TLIMHA/G |
| VPg | 5933–6526 | 594/198 | QALASE/T |
| NIa-Pro | 6527–7240 | 714/238 | HGAEVQ/H |
| NIb | 7241–8746 | 1506/502 | ATVDGQ/G |
| CP | 8747–9592 | 846/281 | - |
| P3N-PIPO | 2684–3505 | 824/274 | - |
| 3′-UTR | 9593–9808 | 216/- |
| Recombinant | Major Parent | Minor Parent | Region | Detection Methods | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| R | G | B | M | C | S | T | ||||
| TPT | JP2 | Unknown | 6602–7286 | + | + | + | + | + | + | + |
| IND369 | IND671 | TPT | 4122–4589 | + | − | + | + | + | − | + |
| IND369 | IND671 | INDR-71 | 5333–6340 | + | + | + | + | + | + | + |
| IND5268 | Unknown | IND671 | 4818–5253 | + | + | + | + | + | + | − |
| IND5268 | INDR-71 | IR-Khuz6 | 5667–6992 | + | − | + | + | + | − | + |
| IND5268 | IND67 | YN-21 | 8763–9001 | + | + | + | + | + | − | + |
| PAK | IR-Khuz6 | IND671 | 1454–4156 | + | − | + | + | + | + | + |
| IR-Khuz6 | IR-Khua57 | PAK | 8318–9386 | + | − | + | + | + | − | + |
| INDR-71 | IND369 | IR-Khuz6 | 52–774 | + | − | + | + | + | + | + |
| Recombinant | Parent (Major × Minor) | Region | Detection Methods | ||||||
|---|---|---|---|---|---|---|---|---|---|
| R | G | B | M | C | S | T | |||
| PAK | IR-Khuz6 × IND671 | CI | + | − | + | + | + | + | + |
| IND369 | IND671 × INDR-71 | CI | + | + | + | + | + | + | + |
| INDR-71 | IND5268 × Unknown | CI | − | − | + | + | + | + | + |
| IND369 | IND671 × MYA-Formosa | CP | + | + | + | + | + | + | + |
| INDR-71 | IND671 × Unknown | P1 | + | + | + | + | + | + | + |
| Group | I | II |
|---|---|---|
| I | 0.03 | 0.3 |
| II | 0.3 | 0.10 |
| Protein | dN/dS | Sites under Positive Selection | Protein | dN/dS | Sites under Positive Selection |
|---|---|---|---|---|---|
| P1 | 0.0938 | 1 | VPg | 0.0244 | 0 |
| HC-Pro | 0.0476 | 0 | NIa-Pro | 0.0681 | 0 |
| P3 | 0.0767 | 0 | NIb | 0.0367 | 0 |
| 6K1 | 0.0258 | 0 | CP | 0.0763 | 0 |
| CI | 0.029 | 0 | P3N-PIPO | 0.112 | 1 |
| 6K2 | 0.0013 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, X.-L.; Mai, Z.-Y.; Wei, K.-J.; Huang, Y.-J.; Shan, H.-L.; Cheng, D.-J. Complete Genomic Sequence Analysis of a Sugarcane Streak Mosaic Virus Isolate from Yunnan Province of China. Genes 2023, 14, 1713. https://doi.org/10.3390/genes14091713
Su X-L, Mai Z-Y, Wei K-J, Huang Y-J, Shan H-L, Cheng D-J. Complete Genomic Sequence Analysis of a Sugarcane Streak Mosaic Virus Isolate from Yunnan Province of China. Genes. 2023; 14(9):1713. https://doi.org/10.3390/genes14091713
Chicago/Turabian StyleSu, Xiao-Ling, Zhong-Yue Mai, Kun-Jiang Wei, Yang-Jian Huang, Hong-Li Shan, and De-Jie Cheng. 2023. "Complete Genomic Sequence Analysis of a Sugarcane Streak Mosaic Virus Isolate from Yunnan Province of China" Genes 14, no. 9: 1713. https://doi.org/10.3390/genes14091713
APA StyleSu, X.-L., Mai, Z.-Y., Wei, K.-J., Huang, Y.-J., Shan, H.-L., & Cheng, D.-J. (2023). Complete Genomic Sequence Analysis of a Sugarcane Streak Mosaic Virus Isolate from Yunnan Province of China. Genes, 14(9), 1713. https://doi.org/10.3390/genes14091713