
Snijders Blok–Campeau Syndrome: Description of 20 Additional Individuals with Variants in CHD3 and Literature Review

 , , , , , , , , , ,
, , , , , , , , , ,  , , , add
Show full author list
, , , add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. Genetic Analysis
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Snijders Blok, L.; Rousseau, J.; Twist, J.; Ehresmann, S.; Takaku, M.; Venselaar, H.; Rodan, L.H.; Nowak, C.B.; Douglas, J.; Swoboda, K.J.; et al. CHD3 Helicase Domain Mutations Cause a Neurodevelopmental Syndrome with Macrocephaly and Impaired Speech and Language. Nat. Commun. 2018, 9, 4619. [Google Scholar] [CrossRef] [PubMed]
- Drivas, T.G.; Li, D.; Nair, D.; Alaimo, J.T.; Alders, M.; Altmüller, J.; Barakat, T.S.; Bebin, E.M.; Bertsch, N.L.; Blackburn, P.R.; et al. A Second Cohort of CHD3 Patients Expands the Molecular Mechanisms Known to Cause Snijders Blok-Campeau Syndrome. Eur. J. Hum. Genet. 2020, 28, 1422–1431. [Google Scholar] [CrossRef] [PubMed]
- Coursimault, J.; Lecoquierre, F.; Saugier-Veber, P.; Drouin-Garraud, V.; Lechevallier, J.; Boland, A.; Deleuze, J.-F.; Frebourg, T.; Nicolas, G.; Brehin, A.-C. Hypersociability Associated with Developmental Delay, Macrocephaly and Facial Dysmorphism Points to CHD3 Mutations. Eur. J. Med. Genet. 2021, 64, 104166. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, M.; Ishikawa, A.; Miyazaki, S.; Tsuzuki, A.; Saito, S.; Niihori, T.; Sakurai, A. A de Novo CHD3 Variant in a Child with Intellectual Disability, Autism, Joint Laxity, and Dysmorphisms. Brain Dev. 2021, 43, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.-Y. Snijders Blok-Campeau Syndrome Caused by CHD3 Gene Mutation: A Case Report. Zhongguo Dang Dai Er Ke Za Zhi Chin. J. Contemp. Pediatr. 2021, 23, 965–968. [Google Scholar]
- LeBreton, L.; Allain, E.P.; Parscan, R.C.; Crapoulet, N.; Almaghraby, A.; Ben Amor, M. A Novel CHD3 Variant in a Patient with Central Precocious Puberty: Expanded Phenotype of Snijders Blok-Campeau Syndrome? Am. J. Med. Genet. A 2023, 191, 1065–1069. [Google Scholar] [CrossRef]
- Cardoso, A.R.; Lopes-Marques, M.; Oliveira, M.; Amorim, A.; Prata, M.J.; Azevedo, L. Genetic Variability of the Functional Domains of Chromodomains Helicase DNA-Binding (CHD) Proteins. Genes 2021, 12, 1827. [Google Scholar] [CrossRef] [PubMed]
- Van der Spek, J.; den Hoed, J.; Snijders Blok, L.; Dingemans, A.J.M.; Schijven, D.; Nellaker, C.; Venselaar, H.; Astuti, G.D.N.; Barakat, T.S.; Bebin, E.M.; et al. Inherited Variants in CHD3 Show Variable Expressivity in Snijders Blok-Campeau Syndrome. Genet. Med. 2022, 24, 1283–1296. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Tatton-Brown, K.; Rahman, N. Sotos Syndrome. Eur. J. Hum. Genet. 2007, 15, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Priolo, M.; Schanze, D.; Tatton-Brown, K.; Mulder, P.A.; Tenorio, J.; Kooblall, K.; Acero, I.H.; Alkuraya, F.S.; Arias, P.; Bernardini, L.; et al. Further Delineation of Malan Syndrome. Hum. Mutat. 2018, 39, 1226–1237. [Google Scholar] [CrossRef]
- Tenorio, J.; Mansilla, A.; Valencia, M.; Martínez-Glez, V.; Romanelli, V.; Arias, P.; Castrejón, N.; Poletta, F.; Guillén-Navarro, E.; Gordo, G.; et al. A New Overgrowth Syndrome Is Due to Mutations in RNF125. Hum. Mutat. 2014, 35, 1436–1441. [Google Scholar] [CrossRef] [PubMed]
- Tenorio, J.; Arias, P.; Martínez-Glez, V.; Santos, F.; García-Miñaur, S.; Nevado, J.; Lapunzina, P. Simpson-Golabi-Behmel Syndrome Types I and II. Orphanet J. Rare Dis. 2014, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Torchy, M.P.; Hamiche, A.; Klaholz, B.P. Structure and Function Insights into the NuRD Chromatin Remodeling Complex. Cell. Mol. Life Sci. 2015, 72, 2491–2507. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Gao, S.; Schafer, C.; Colijn, S.; Muthukumar, V.; Griffin, C.T. The Chromatin-Remodeling Enzyme CHD3 Plays a Role in Embryonic Viability but Is Dispensable for Early Vascular Development. PLoS ONE 2020, 15, e0235799. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Ou, X.; Li, J. Mechanistic Insight on General Protein-Binding Ability of ATP and the Impacts of Arginine Residues. J. Phys. Chem. B 2022, 126, 4647–4658. [Google Scholar] [CrossRef]


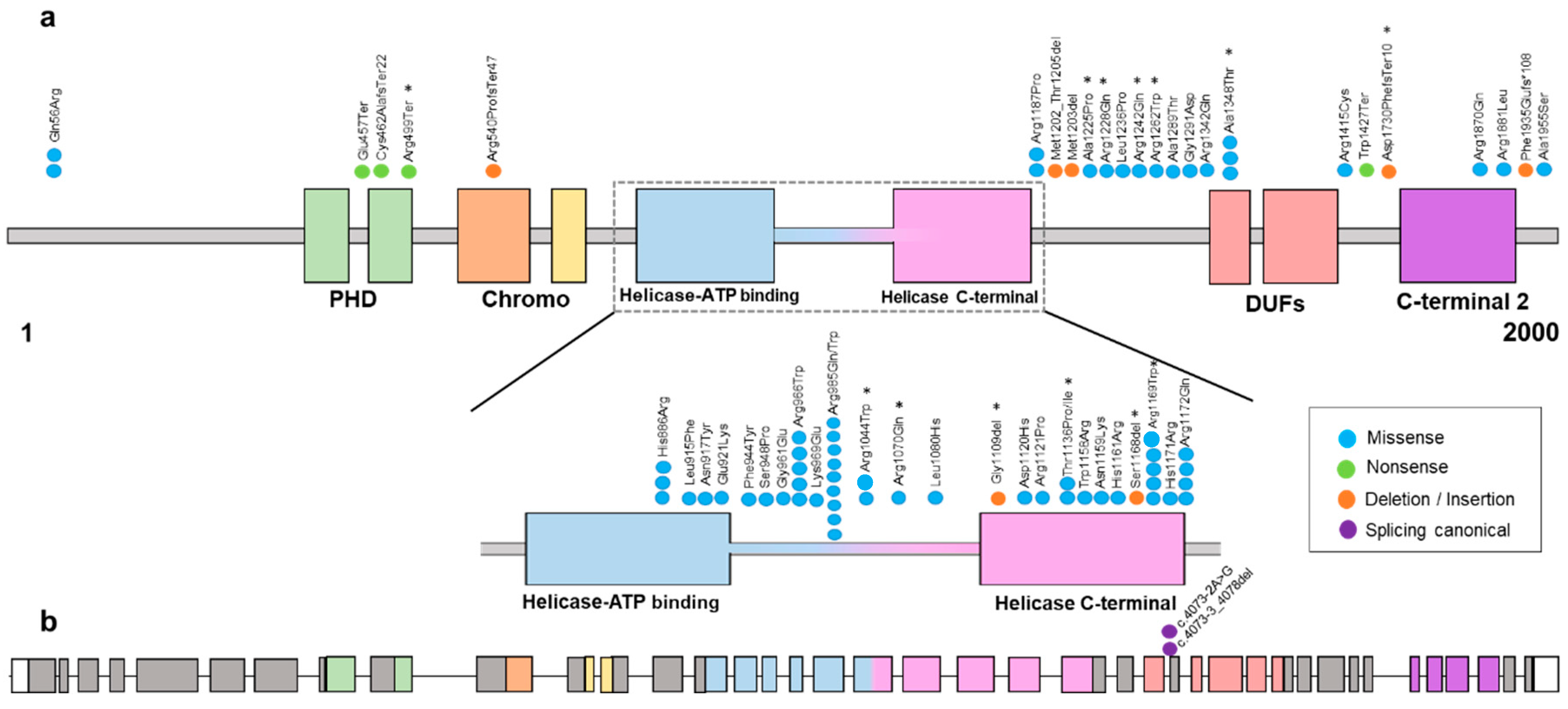{kind=link}
| Frequency | Snijders Blok–Campeau Patients | Drivas et al., 2020 | Mizukami et al., 2021 | Xi-Yong, 2021 | Coursimault et al., 2021 | Snijders Blok et al., 2018 | LeBreton et al., 2022 | This Study (n = 20) | Total (n = 83) | Percentage (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| HP:0001263 Global developmental delay | 24 | 1 | 1 | 35 | 1 | 19 | 81 | 98% | ||
| >75% | HP:0002167 Speech disorder/delay | 24 | 1 | 1 | 1 | 33 | 1 | 15 | 76 | 92% |
| HP:0001249 Intellectual disability | 20 | 1 | 1 | 1 | 27 | 11 | 61 | 73% | ||
| HP:0001252 Hypotonia | 22 | 1 | 1 | 1 | 21 | 13 | 59 | 71% | ||
| HP:0000316 Hypertelorism | 13 | 1 | 1 | 24 | 13 | 52 | 63% | |||
| 51–74% | HP:0000504 Abnormality of vision | 18 | 1 | 1 | 23 | 3 | 46 | 55% | ||
| HP:0000256 Macrocephaly | 10 | 1 | 1 | 19 | 15 | 46 | 55% | |||
| HP:0000244 Prominent forehead | 1 | 28 | 14 | 43 | 52% | |||||
| HP:0002011 Abnormality of CNS | 9 | 1 | 16 | 9 | 35 | 42% | ||||
| HP:0002007 Frontal bossing | 13 | 11 | 1 | 10 | 35 | 42% | ||||
| HP:0000307 Pointed chin | 12 | 1 | 1 | 13 | 27 | 33% | ||||
| HP:0000431 Wide nasal bridge | 17 | 1 | 8 | 26 | 31% | |||||
| 26–50% | HP:0000219 Thin upper lips | 17 | 9 | 26 | 31% | |||||
| HP:0001388 Joint laxity | 1 | 1 | 1 | 12 | 10 | 25 | 30% | |||
| HP:0000717 Autism spectrum disorder | 9 | 1 | 1 | 9 | 1 | 2 | 23 | 28% | ||
| HP:0000486 Strabismus | 6 | 1 | 1 | 1 | 10 | 4 | 23 | 28% | ||
| HP:0000293 Full cheeks | 13 | 1 | 8 | 22 | 27% | |||||
| HP:0005338 Sparse lateral eyebrow | 12 | 1 | 1 | 5 | 19 | 23% | ||||
| HP:0001270 Motor delay | 1 | 1 | 17 | 19 | 23% | |||||
| HP:0000369 Low-set ears | 8 | 1 | 1 | 1 | 1 | 5 | 17 | 20% | ||
| HP:0000490 Deep-set eyes | 13 | 1 | 2 | 16 | 19% | |||||
| HP:0031936 Delayed walking | 16 | 16 | 19% | |||||||
| HP:0045025 Narrow palpebral fissure | 10 | 1 | 1 | 3 | 15 | 18% | ||||
| HP:0000506 Telecanthus | 10 | 1 | 1 | 1 | 13 | 16% | ||||
| HP:0000448 Prominent nose | 6 | 1 | 1 | 4 | 12 | 14% | ||||
| HP:0012371 Mid-face hypoplasia | 9 | 1 | 2 | 12 | 14% | |||||
| HP:0001388 Abnormality of the male genitalia | 1 | 6 | 5 | 12 | 14% | |||||
| HP:0001627 Congenital heart defects | 5 | 3 | 3 | 11 | 13% | |||||
| HP:0001250 Seizures | 5 | 4 | 2 | 11 | 13% | |||||
| HP:0008872 Feeding difficulties in infancy | 10 | 1 | 11 | 13% | ||||||
| HP:0000358 Posteriorly rotated ears | 9 | 1 | 10 | 12% | ||||||
| HP:0000337 Broad forehead | 10 | 10 | 12% | |||||||
| HP:0000286 Epicanthus | 10 | 10 | 12% | |||||||
| HP:0006349 Absent teeth | 5 | 2 | 1 | 8 | 10% | |||||
| HP:0007018 Attention deficit hyperactivity disorder | 1 | 7 | 8% | |||||||
| HP:0100790 Hernia | 5 | 6 | 6 | 7% | ||||||
| <25% | HP:0000455 Broad nasal tip | 6 | 1 | 6 | 7% | |||||
| HP:0000164 Dental abnormalities | 1 | 5 | 6 | 7% | ||||||
| HP:0000733 Stereotypic behavior | 1 | 4 | 5 | 6% | ||||||
| HP:0000218 High palate | 5 | 5 | 6% | |||||||
| HP:0001760 Foot deformities | 4 | 4 | 5% | |||||||
| HP:0002119 Ventriculomegaly | 1 | 3 | 4 | 5% | ||||||
| HP:0005280 Depressed nasal root | 4 | 4 | 5% | |||||||
| HP:0001041 Blushed cheeks | 3 | 3 | 4% | |||||||
| HP:0000365 Hearing impairment | 3 | 3 | 4% | |||||||
| HP:0001260 Dysarthria | 3 | 3 | 4% | |||||||
| HP:0000252 Microcephaly | 2 | 1 | 3 | 4% | ||||||
| HP:0002317 Unsteady gait | 2 | 2 | 2% | |||||||
| HP:0000957 Cafe-au-lait spot /HP:0007565 Multiple cafe-au-lait spots | 2 | 2 | 2% | |||||||
| HP:0045025 Narrow palpebral fissure | 10 | 3 | 2 | 2% | ||||||
| HP:0040082 Happy demeanor | 1 | 1 | 2 | 2% | ||||||
| HP:0000753 Autism with high cognitive abilities | 1 | 1 | 1% | |||||||
| HP:0000077 Abnormality of the kidney | 1 | 1 | 1% | |||||||
| HP:0000664 Synophris | 1 | 1 | 1% | |||||||
| HP:0000322 Short philtrum | 1 | 1 | 1% | |||||||
| HP:0000160 Narrow mouth | 1 | 1 | 1% | |||||||
| HP:0004442 Sagittal craniosynostosis | 1 | 1 | 1% | |||||||
| HP:0001047 Pes planus | 1 | 1 | 1% | |||||||
| HP:0001047 Atopic dermatitis | 1 | 1 | 1% | |||||||
| HP:0045074 Thin eyebrow | 1 | 1 | 1% |
| Family | Proband | Genomic Coordinate (hg38) | cDNA and Protein Location | Exon/Intron | Mutation Type | Zygosity | Inheritance | Population Frequency # | CADD | REVEL | ACMG Prediction |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | chr17:7811001 | NM_001005271.3:c.5184_5185del (p.Asp1730PhefsTer10) | 33 | Frameshift | Heterozygous | De novo | - | 35 | - | Likely Pathogenic |
| 2 | 2 | chr17:7804024 | NM_001005271.3:c.3130C>T (p.Arg1044Trp) | 18 | Missense | Heterozygous | De novo | - | 29.8 | - | Pathogenic |
| 3 | 3 | chr17:7804650 | NM_001005273.3:c.3209G>A (p.Arg1070Gln) | 20 | Missense | Heterozygous | De novo | 0.0000159 | 24.9 | 0.161 | Likely Pathogenic |
| 4 | 4 | chr17:7806590 | NM_001005271.3:c.3673G>C (p.Ala1225Pro) | 22 | Missense | Heterozygous | De novo | - | 33 | - | Pathogenic |
| 5 | 5 | chr17:7805996 | NM_001005271.3:c.3325_3327delGGT (p.Tyr1109del) | 20 | In frame deletion | Heterozygous | De novo | - | - | - | Likely Pathogenic |
| 6 | 6 | chr17:7806642 | NM_001005271.3:c.3725G>A (p.Arg1242Gln) | 23 | Missense | Heterozygous | Inherited from father | - | 32 | 0.947 | Likely Pathogenic |
| 7 | 7 | chr17:7806600 | NM_001005271.3:c.3683G>A (p.Arg1228Gln) | 23 | Missense | Heterozygous | De novo | - | 31 | 0.957 | Pathogenic |
| 8 | 8 | chr17:7903278 | NM_001005273.3:c.3502_3504del (p.Ser1168del) | 23 | In frame deletion | Heterozygous | De novo | - | - | - | Likely Pathogenic |
| 9 | 9 | chr17:7902972 | NM_001005273.3:c.3406A>C (p.Thr1136Pro) | 22 | Missense | Heterozygous | De novo | - | 29.9 | 0.966 | Likely Pathogenic |
| 10 | 10 | chr17:7895142 | NM_001005273.3:c.1495C>T (p.Arg499Ter) | 9 | Nonsense | Heterozygous | De novo | - | - | - | Pathogenic |
| 11 | 11 | chr17:7903962 | NM_001005273.3:c.3865G>A (p.Ala1289Thr) | 24 | Missense | Heterozygous | Unknown | - | 29.1 | 0.831 | Likely Pathogenic |
| 12 | 12 | chr17:7903962 | NM_001005271.3:c.4042G>A (p.Ala1348Thr) | 24 | Missense | Heterozygous | Inherited from mother | - | 29.1 | 0.831 | Likely Pathogenic |
| 12 | 13 | chr17:7903962 | NM_001005271.3:c.4042G>A (p.Ala1348Thr) | 24 | Missense | Heterozygous | Inherited from mother | - | 29.1 | 0.831 | Likely Pathogenic |
| 12 | 14 | chr17:7903962 | NM_001005271.3:c.4042G>A (p.Ala1348Thr) | 24 | Missense | Heterozygous | Inherited from mother | - | 29.1 | 0.831 | Likely Pathogenic |
| 13 | 15 | chr17:7900707 | NM_001005273.3:c.2954G>A (p.Arg985Gln) | 18 | Missense | Heterozygous | De novo | - | 32 | 0.777 | Pathogenic |
| 14 | 16 | chr17:7903281 | NM_001005273.3:c.3505C>T p.Arg1169Trp | 23 | Missense | Heterozygous | De novo | - | 25.7 | 0.899 | Pathogenic |
| 15 | 17 | chr17:7903317 | NM_001005273.3:c.3541A>T p.Ile1181Phe | 23 | Missense | Heterozygous | De novo | - | 28.4 | 0.904 | Likely Pathogenic |
| 15 | 18 | chr17:7903317 | NM_001005273.3:c.3541A>T p.Ile1181Phe | 23 | Missense | Heterozygous | De novo | - | 28.4 | 0.904 | Likely Pathogenic |
| 16 | 19 | chr17:7903282 | NM_001005273.3:c.3506G>A p.Arg1169Gln | 23 | Missense | Heterozygous | De novo | - | 31 | 0.96 | Pathogenic |
| 17 | 20 | chr17:7804024 | NM_001005271.3:c.3130C>T (p.Arg1044Trp) | 18 | Missense | Heterozygous | De novo | - | 29.8 | - | Pathogenic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pascual, P.; Tenorio-Castano, J.; Mignot, C.; Afenjar, A.; Arias, P.; Gallego-Zazo, N.; Parra, A.; Miranda, L.; Cazalla, M.; Silván, C.; et al. Snijders Blok–Campeau Syndrome: Description of 20 Additional Individuals with Variants in CHD3 and Literature Review. Genes 2023, 14, 1664. https://doi.org/10.3390/genes14091664
Pascual P, Tenorio-Castano J, Mignot C, Afenjar A, Arias P, Gallego-Zazo N, Parra A, Miranda L, Cazalla M, Silván C, et al. Snijders Blok–Campeau Syndrome: Description of 20 Additional Individuals with Variants in CHD3 and Literature Review. Genes. 2023; 14(9):1664. https://doi.org/10.3390/genes14091664
Chicago/Turabian StylePascual, Patricia, Jair Tenorio-Castano, Cyril Mignot, Alexandra Afenjar, Pedro Arias, Natalia Gallego-Zazo, Alejandro Parra, Lucia Miranda, Mario Cazalla, Cristina Silván, and et al. 2023. "Snijders Blok–Campeau Syndrome: Description of 20 Additional Individuals with Variants in CHD3 and Literature Review" Genes 14, no. 9: 1664. https://doi.org/10.3390/genes14091664
APA StylePascual, P., Tenorio-Castano, J., Mignot, C., Afenjar, A., Arias, P., Gallego-Zazo, N., Parra, A., Miranda, L., Cazalla, M., Silván, C., Heron, D., Keren, B., Popa, I., Palomares, M., Rikeros, E., Ramos, F. J., Almoguera, B., Ayuso, C., Swafiri, S. T., ... Lapunzina, P. (2023). Snijders Blok–Campeau Syndrome: Description of 20 Additional Individuals with Variants in CHD3 and Literature Review. Genes, 14(9), 1664. https://doi.org/10.3390/genes14091664