

Analyzes In Silico Indicate the lncRNAs MIR31HG and LINC00939 as Possible Epigenetic Inhibitors of the Osteogenic Differentiation in PDLCs
 ,
,  ,
,
Abstract
:1. Introduction
2. Material and Methods
2.1. Cell Acquisition and Culture
2.2. Osteogenic Stimulation
- (i)
- l-DMEM: l-PDLCs cultivated in DMEM, standard medium.
- (ii)
- l-OM: l-PDLCs cultivated in OM, osteogenic medium.
- (iii)
- h-DMEM: h-PDLCs cultivated in DMEM, standard medium.
- (iv)
- h-OM: h-PDLCs cultivated in OM, osteogenic medium.
2.3. Assay for Transposase-Accessible Chromatin Using Sequencing (ATAC-Seq)
2.4. Global DNA Methylation Analysis (Methylome)
DNA Isolation and Oxidative Bisulfite Conversion
2.5. RNA Sequencing (RNA-SEQ)
2.5.1. I. RNA Extraction
2.5.2. II. RNA Sequencing
2.6. Bioinformatics Analysis
2.6.1. III. ATAC-Seq
2.6.2. IV. DNA Methylome
2.6.3. RNA-Seq
2.6.4. Selection of lncRNAs and Prediction Analyses
2.7. PCR Analysis
2.8. Statistical Analysis of the PCR Data
3. Results
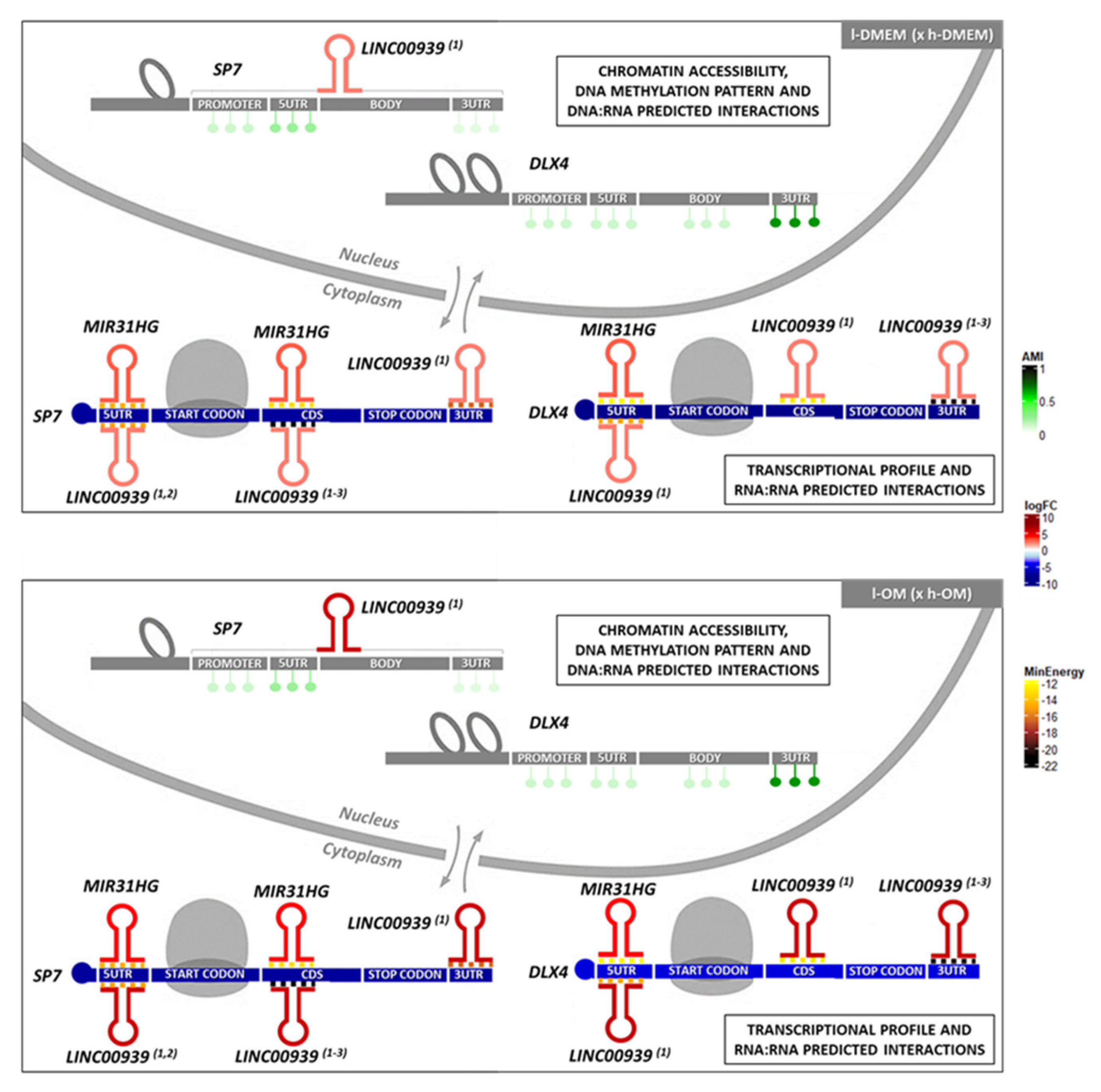
3.1. l-PDLCs Show More Accessible Chromatin Regions in SP7 and DLX4 Genes than h-PDLCs
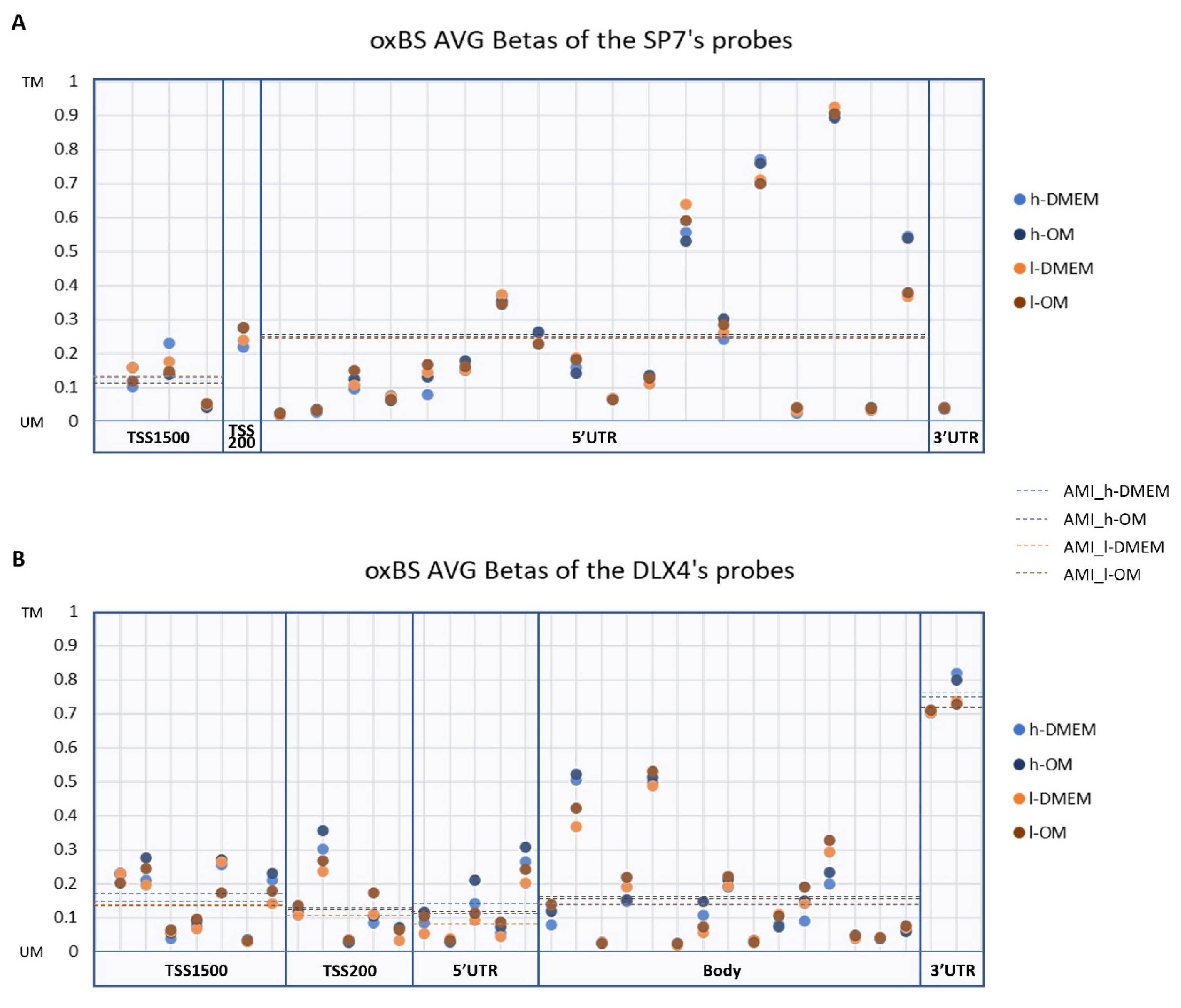
3.2. DNA Methylation Patterns amongst l- and h-PDLCS Are Similar for SP7 and DLX4
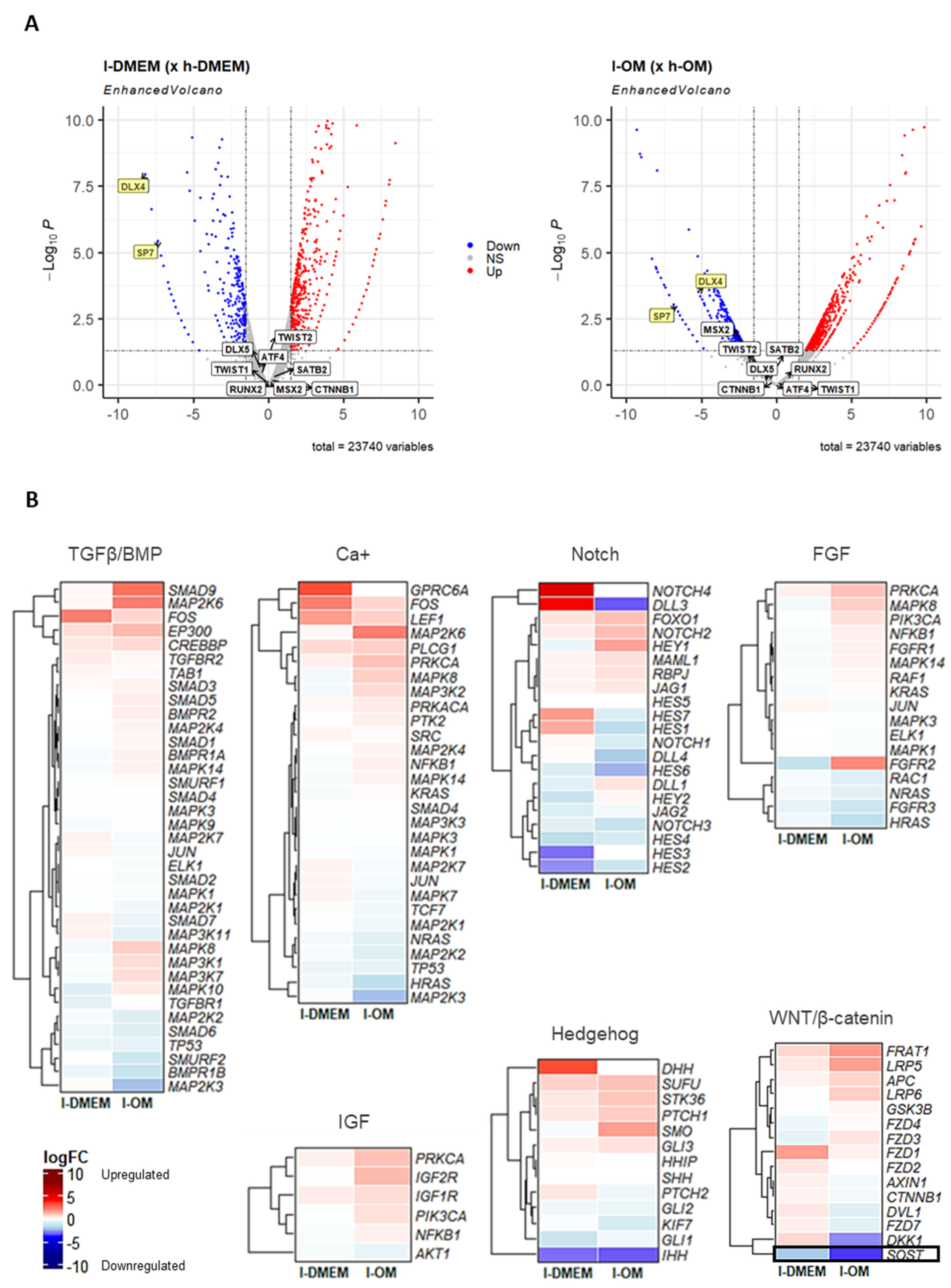
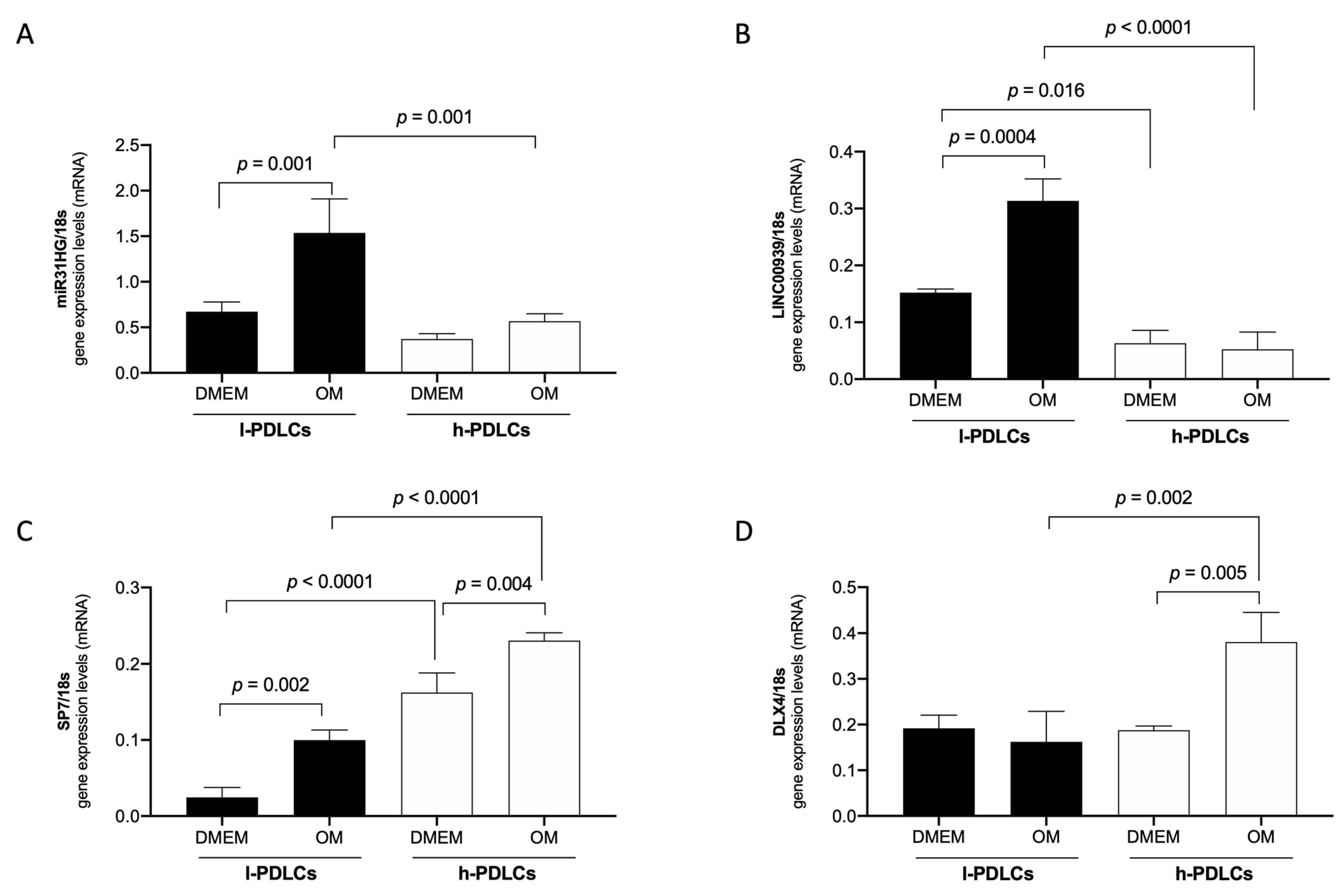
3.3. The lncRNAs LINC00939 and MIR31HG Are Upregulated in l-PDLCs
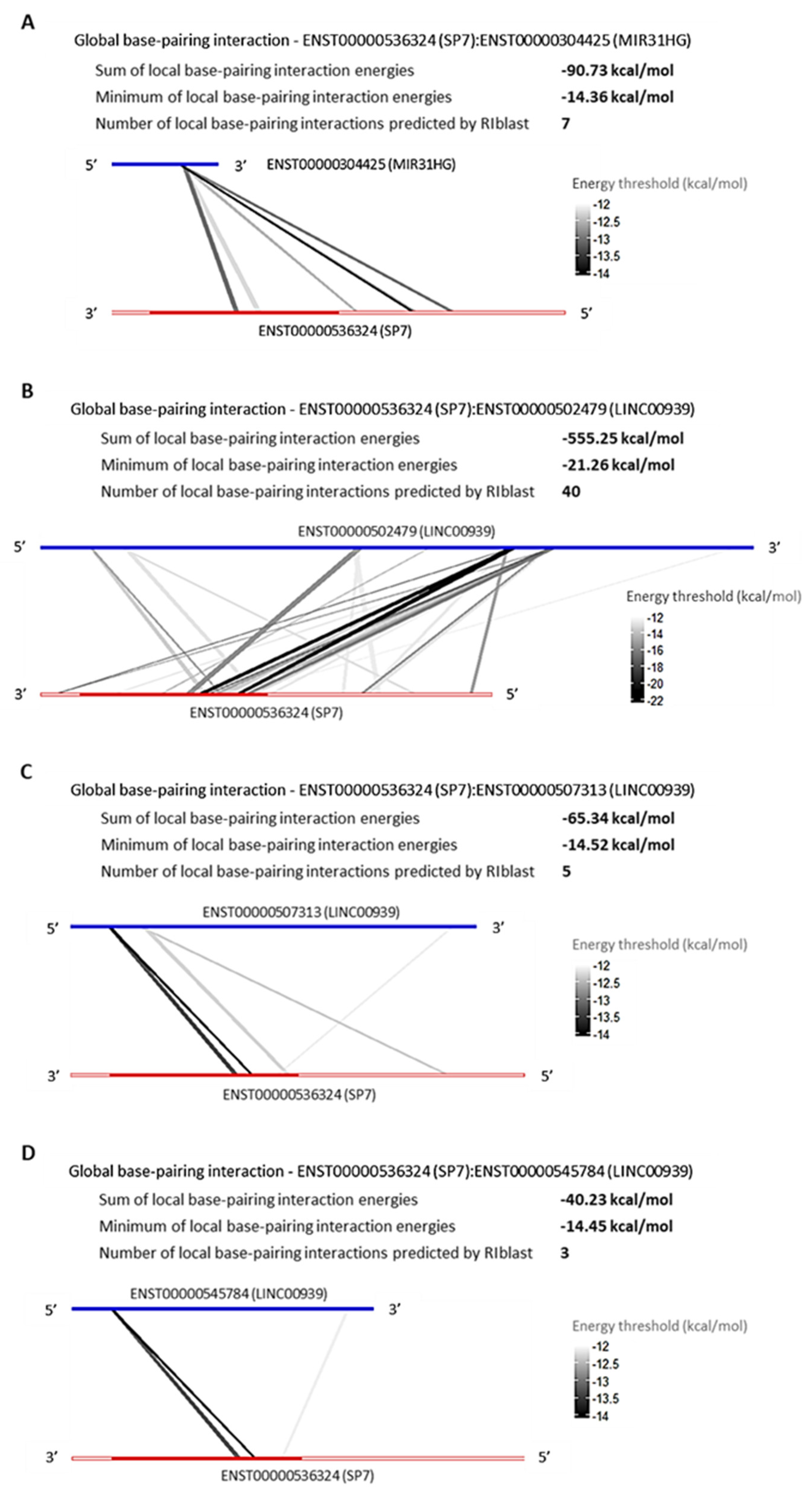
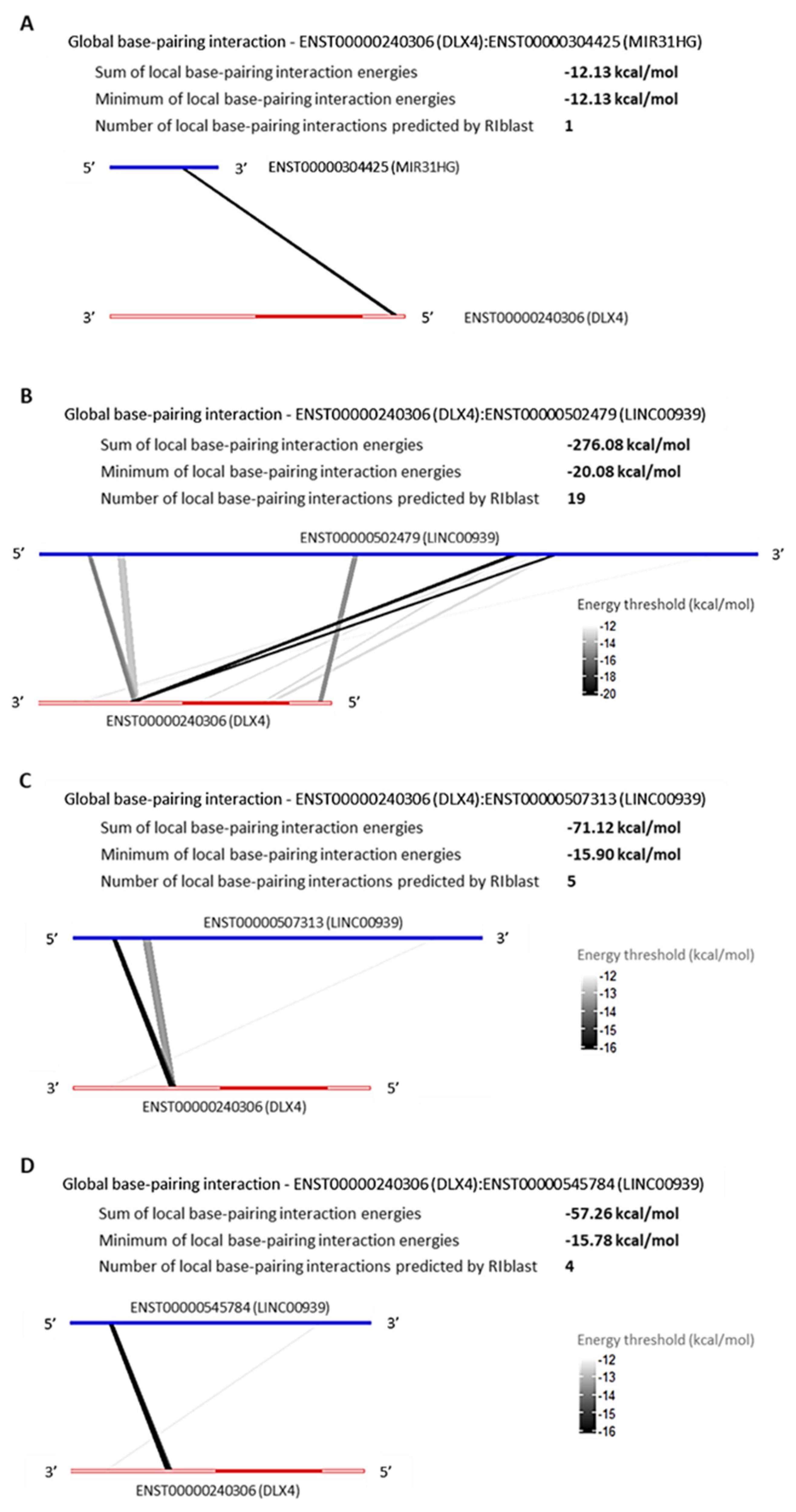
LncRNAs, MIR31HG and LINC00939 Are Predicted to Interact with SP7 and DLX4 by RNA:RNA Base-Pairing
3.4. Machine Learning Program Predicted Potential for DNA:RNA Triplex-Forming for the SP7 DNA Site and for the lncRNA LINC00939
3.5. MIR31HG and LINC00939 Transcripts Are Upregulated while SP7 and DLX4 Are Downregulated in l-PDLCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hmadcha, A.; Martin-Montalvo, A.; Gauthier, B.R.; Soria, B.; Capilla-Gonzalez, V. Therapeutic potential of mesenchymal stem cells for cancer therapy. Front. Bioeng. Biotechnol. 2020, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Meirelles, L.D.S.; Chagastelles, P.C.; Nardi, N.B. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J. Cell. Sci. 2006, 119, 2204–2213. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, K.M.; Diogenes, A.; Teixeira, F.B. Treatment options: Biological basis of regenerative endodontic procedures. Pediatr. Dent. 2013, 35, 129–140. [Google Scholar] [CrossRef]
- Dominici, M.L.B.K.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells: The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Salgado, A.J.; Reis, R.L.; Sousa, N.; Gimble, J.M. Adipose tissue derived stem cells secretome: Soluble factors and their roles in regenerative medicine. Curr. Stem Cell Res. Ther. 2010, 5, 103–110. [Google Scholar] [CrossRef]
- Jiang, Y.; Jahagirdar, B.N.; Reinhardt, R.L.; Schwartz, R.E.; Keene, C.D.; Ortiz-Gonzalez, X.R.; Reyes, M.; Lenvik, T.; Lund, T.; Blackstad, M.; et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 2002, 418, 41–49. [Google Scholar] [CrossRef]
- Sevari, S.P.; Ansari, S.; Moshaverinia, A. A narrative overview of utilizing biomaterials to recapitulate the salient regenerative features of dental-derived mesenchymal stem cells. Int. J. Oral Sci. 2021, 13, 22. [Google Scholar] [CrossRef]
- Saito, M.T.; Salmon, C.R.; Amorim, B.R.; Ambrosano, G.M.; Casati, M.Z.; Sallum, E.A.; Nociti, F.H., Jr.; Silvério, K.G. Characterization of highly osteoblast/cementoblast cell clones from a CD105-enriched periodontal ligament progenitor cell population. J. Periodontol. 2014, 85, e205–e211. [Google Scholar] [CrossRef]
- Assis, R.I.; Feltran, G.D.S.; Silva, M.E.S.; do Rosário Palma, I.C.; Rovai, E.S.; de Miranda, T.B.; Ferreira, M.R.; Zambuzzi, W.F.; Birbrair, A.; Andia, D.C.; et al. Non-coding RNAs repressive role in post-transcriptional processing of RUNX2 during the acquisition of the osteogenic phenotype of periodontal ligament mesenchymal stem cells. Dev. Biol. 2021, 470, 37–48. [Google Scholar] [CrossRef]
- Assis, R.I.; Schmidt, A.G.; Racca, F.; da Silva, R.A.; Zambuzzi, W.F.; Silvério, K.G.; Nociti, F.H., Jr.; Pecorari, V.G.; Wiench, M.; Andia, D.C. DNMT1 inhibitor restores RUNX2 expression and mineralization in periodontal ligament cells. DNA Cell Biol. 2021, 40, 662–674. [Google Scholar] [CrossRef]
- Ferreira, R.S.; Assis, R.I.; Feltran, G.D.S.; do Rosário Palma, I.C.; Françoso, B.G.; Zambuzzi, W.F.; Andia, D.C.; da Silva, R.A. Genome-wide DNA (hydroxy) methylation reveals the individual epigenetic landscape importance on osteogenic phenotype acquisition in periodontal ligament cells. J. Periodontol. 2022, 93, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Assis, R.I.; Racca, F.; Ferreira, R.S.; Ruiz, K.G.; da Silva, R.A.; Clokie, S.J.; Wiench, M.; Andia, D.C. Osteogenic commitment of human periodontal ligament cells is predetermined by methylation, chromatin accessibility and expression of key transcription factors. Cells 2022, 11, 1126. [Google Scholar] [CrossRef] [PubMed]
- Stein, G.S.; Zaidi, S.K.; Stein, J.L.; Lian, J.B.; Van Wijnen, A.J.; Montecino, M.; Young, D.W.; Javed, A.; Pratap, J.; Choi, J.; et al. Transcription-factor-mediated epigenetic control of cell fate and lineage commitment. Biochem. Cell Biol. 2009, 87, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Morrison, O.; Thakur, J. Molecular complexes at euchromatin, heterochromatin and centromeric chromatin. Int. J. Mol. Sci. 2021, 22, 6922. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Lambert, S.A.; Jolma, A.; Campitelli, L.F.; Das, P.K.; Yin, Y.; Albu, M.; Chen, X.; Taipale, J.; Hughes, T.R.; Weirauch, M.T. The human transcription factors. Cell 2018, 172, 650–665. [Google Scholar] [CrossRef]
- Pratt, A.J.; MacRae, I.J. The RNA-induced silencing complex: A versatile gene-silencing machine. J. Biol. Chem. 2009, 284, 17897–17901. [Google Scholar] [CrossRef]
- Kunkler, C.N.; Hulewicz, J.P.; Hickman, S.C.; Wang, M.C.; McCown, P.J.; Brown, J.A. Stability of an RNA• DNA–DNA triple helix depends on base triplet composition and length of the RNA third strand. Nucleic Acids Res. 2019, 47, 7213–7222. [Google Scholar] [CrossRef]
- Li, Y.; Syed, J.; Sugiyama, H. RNA-DNA triplex formation by long noncoding RNAs. Cell Chem. Biol. 2016, 23, 1325–1333. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Silvério, K.G.; Rodrigues, T.L.; Coletta, R.D.; Benevides, L.; Da Silva, J.S.; Casati, M.Z.; Sallum, E.A.; Nociti, F.H., Jr. Mesenchymal stem cell properties of periodontal ligament cells from deciduous and permanent teeth. J. Periodontol. 2010, 81, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- UCSC: Genome Browser. University of California Santa Cruz. Available online: https://genome.ucsc.edu/ (accessed on 28 May 2023).
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. ATAC-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef]
- Corces, M.R.; Buenrostro, J.D.; Wu, B.; Greenside, P.G.; Chan, S.M.; Koenig, J.L.; Snyder, M.P.; Pritchard, J.K.; Kundaje, A.; Greenleaf, W.J.; et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016, 48, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Illumina: GenomeStudio Software. Available online: https://www.illumina.com/techniques/microarrays/array-data-analysis-experimental-design/genomestudio.html (accessed on 28 May 2023).
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef]
- Wickham, H.; François, R.; Henry, L.; Müller, K. dplyr: A Grammar of Data Manipulation. Available online: https://github.com/tidyverse/dplyr (accessed on 28 May 2023).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.A.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-ready volcano plots with enhanced coloring and labeling. In R Package Version 1.8.0; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Fukunaga, T.; Iwakiri, J.; Ono, Y.; Hamada, M. LncRRIsearch: A web server for lncRNA-RNA interaction prediction integrated with tissue-specific expression and subcellular localization data. Front. Genet. 2019, 10, 462. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, C.J.; Schmidt, R.; Kanitz, A.; Artimo, P.; Gruber, A.J.; Zavolan, M. PolyASite 2.0: A consolidated atlas of polyadenylation sites from 3′ end sequencing. Nucleic Acids Res. 2020, 48, D174–D179. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Long, Y.; Kwoh, C.K. Deep learning based DNA: RNA triplex forming potential prediction. BMC Bioinform. 2020, 21, 522. [Google Scholar] [CrossRef]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Assis, R.I.; Wiench, M.; Silverio, K.G.; da Silva, R.A.; Feltran, G.D.S.; Sallum, E.A.; Casati, M.Z.; Nociti, F.H., Jr.; Andia, D.C. RG108 increases NANOG and OCT4 in bone marrow-derived mesenchymal cells through global changes in DNA modifications and epigenetic activation. PLoS ONE 2018, 13, e0207873. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Tai, P.W.; Wu, H.; Van Wijnen, A.J.; Stein, G.S.; Stein, J.L.; Lian, J.B. Genome-wide DNase hypersensitivity, and occupancy of RUNX2 and CTCF reveal a highly dynamic gene regulome during MC3T3 pre-osteoblast differentiation. PLoS ONE 2017, 12, e0188056. [Google Scholar] [CrossRef]
- Lhoumaud, P.; Sethia, G.; Izzo, F.; Sakellaropoulos, T.; Snetkova, V.; Vidal, S.; Badri, S.; Cornwell, M.; Giammartino, D.C.D.G.; Kim, K.T.; et al. EpiMethylTag: Simultaneous detection of ATAC-seq or ChIP-seq signals with DNA methylation. Genome Biol. 2019, 20, 248. [Google Scholar] [CrossRef]
- Pérez-Campo, F.M.; Santurtún, A.; García-Ibarbia, C.; Pascual, M.A.; Valero, C.; Garcés, C.; Sañudo, C.; Zarrabeitia, M.T.; Riancho, J.A. Osterix and RUNX2 are transcriptional regulators of sclerostin in human bone. Calcif. Tissue Int. 2016, 99, 302–309. [Google Scholar] [CrossRef]
- Ohyama, Y.; Nifuji, A.; Maeda, Y.; Amagasa, T.; Noda, M. Spaciotemporal association and bone morphogenetic protein regulation of sclerostin and osterix expression during embryonic osteogenesis. Endocrinology 2004, 145, 4685–4692. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kfir, E.; Artsi, H.; Levin, A.; Abramowitz, E.; Bajayo, A.; Gurt, I.; Zhong, L.; D’Urso, A.; Toiber, D.; Mostoslavsky, R.; et al. Sirt1 is a regulator of bone mass and a repressor of Sost encoding for sclerostin, a bone formation inhibitor. Endocrinology 2011, 152, 4514–4524. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Yang, Y.S.; Xie, J.; Lee, O.; Kim, J.; Hong, J.; Boldyreff, B.; Filhol, O.; Chun, H.; Greenblatt, M.B.; et al. Regulation of sclerostin by the SIRT1 stabilization pathway in osteocytes. Cell Death Differ. 2022, 29, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Starks, R.R.; Biswas, A.; Jain, A.; Tuteja, G. Combined analysis of dissimilar promoter accessibility and gene expression profiles identifies tissue-specific genes and actively repressed networks. Epigenetics Chromatin 2019, 12, 16. [Google Scholar] [CrossRef]
- Anastasiadi, D.; Esteve-Codina, A.; Piferrer, F. Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics Chromatin 2018, 11, 37. [Google Scholar] [CrossRef]
- Wang, K.; Dai, R.; Xia, Y.; Tian, J.; Jiao, C.; Mikhailova, T.; Zhang, C.; Chen, C.; Liu, C. Spatiotemporal specificity of correlated DNA methylation and gene expression pairs across different human tissues and stages of brain development. Epigenetics 2022, 17, 1110–1127. [Google Scholar] [CrossRef]
- Fukunaga, T.; Hamada, M. RIblast: An ultrafast RNA–RNA interaction prediction system based on a seed-and-extension approach. Bioinformatics 2017, 33, 2666–2674. [Google Scholar] [CrossRef]
- Baglìo, S.R.; Devescovi, V.; Granchi, D.; Baldini, N. MicroRNA expression profiling of human bone marrow mesenchymal stem cells during osteogenic differentiation reveals Osterix regulation by miR-31. Gene 2013, 527, 321–331. [Google Scholar] [CrossRef]
- McCully, M.; Conde, J.; Baptista, P.V.; Mullin, M.; Dalby, M.J.; Berry, C.C. Nanoparticle-antagomiR based targeting of miR-31 to induce osterix and osteocalcin expression in mesenchymal stem cells. PLoS ONE 2018, 13, e0192562. [Google Scholar] [CrossRef]
- Huang, Y.; Zheng, Y.; Xu, Y.; Li, X.; Zheng, Y.; Jia, L.; Li, W. Titanium surfaces functionalized with siMIR31HG promote osteogenic differentiation of bone marrow mesenchymal stem cells. ACS Biomater. Sci. Eng. 2018, 4, 2986–2993. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, W.; Zhu, W.; Dong, J.; Cheng, Y.; Yin, Z.; Shen, F. Mechanisms and functions of long non-coding RNAs at multiple regulatory levels. Int. J. Mol. Sci. 2019, 20, 5573. [Google Scholar] [CrossRef] [PubMed]
- Slotkin, W.; Nishikura, K. Adenosine-to-inosine RNA editing and human disease. Genome Med. 2013, 5, 105. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Maquat, L.E. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature 2011, 470, 284–288. [Google Scholar] [CrossRef]
- Park, E.; Maquat, L.E. Staufen-mediated mRNA decay. Wiley Interdiscip. Rev. RNA 2013, 4, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Browning, D.F.; Busby, S.J. The regulation of bacterial transcription initiation. Nat. Rev. Microbiol. 2004, 2, 57–65. [Google Scholar] [CrossRef]
- Mondal, T.; Subhash, S.; Vaid, R.; Enroth, S.; Uday, S.; Reinius, B.; Mitra, S.; Mohammed, A.; James, A.R.; Hoberg, E.; et al. MEG3 long noncoding RNA regulates the TGF-β pathway genes through formation of RNA–DNA triplex structures. Nat. Commun. 2015, 6, 7743. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.N.; Maione, R. Identification of chromatin binding sites for long noncoding RNAs by chromatin oligo-affinity precipitation (ChOP). In RNA-Chromatin Interactions: Methods and Protocols; Humana: New York, NY, USA, 2020; pp. 17–28. ISBN 978-1-0716-0680-3. [Google Scholar]
- Everaert, C.; Luypaert, M.; Maag, J.L.; Cheng, Q.X.; Dinger, M.E.; Hellemans, J.; Mestdagh, P. Benchmarking of RNA-sequencing analysis workflows using whole-transcriptome RT-qPCR expression data. Sci. Rep. 2017, 7, 1559. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.P.D.C.; Schroen, B.; Kuster, G.M.; Robinson, E.L.; Ford, K.; Squire, I.B.; Heymans, S.; Martelli, F.; Emanueli, C.; Devaux, Y.; et al. Regulatory RNAs in heart failure. Circulation 2020, 141, 313–328. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TRIPLEX DNA SITE PREDICTION | |||||
|---|---|---|---|---|---|
| Gene Symbol | Assembly | Chromosome | Map Location | Score | Triplex-Forming |
| SP7 | GRCh37 | 12 | 53720362:53739099 | 0.68235606 | Triplex |
| DLX4 | GRCh37 | 17 | 48046334:48052321 | 0.13426343 | Nontriplex |
| LNCRNAS TRIPLEX-FORMING POTENTIAL | |||||
|---|---|---|---|---|---|
| Gene Symbol | Transcript ID | Annotation | Exon ID | Score | Triplex-Forming |
| MIR31HG | ENST00000304425.3 | exon | ENSE00001540342 | 0.0 | Nontriplex |
| exon | ENSE00001540341 | 0.0 | Nontriplex | ||
| exon | ENSE00001540339 | 0.0 | Nontriplex | ||
| exon | ENSE00001729409 | 0.0 | Nontriplex | ||
| intron 1 | 0.0 | Nontriplex | |||
| intron 2 | 0.0 | Nontriplex | |||
| intron 3 | 0.0 | Nontriplex | |||
| LINC00939 | ENST00000502479.1 | exon | ENSE00002048191 | 0.7498492 | Triplex |
| exon | ENSE00002044944 | 0.0 | Nontriplex | ||
| exon | ENSE00001441774 | 0.0 | Nontriplex | ||
| exon | ENSE00002049964 | 0.033063784 | Nontriplex | ||
| exon | ENSE00002021220 | 0.30899337 | Nontriplex | ||
| exon | ENSE00002063907 | 0.0 | Nontriplex | ||
| intron 1 | 0.0 | Nontriplex | |||
| intron 2 | 0.0 | Nontriplex | |||
| intron 3 | 0.0 | Nontriplex | |||
| intron 4 | 0.0 | Nontriplex | |||
| intron 5 | 0.0 | Nontriplex | |||
| LINC00939 | ENST00000507313.1 | exon | ENSE00002060110 | 0.0 | Nontriplex |
| exon | ENSE00002044944 | 0.0 | Nontriplex | ||
| exon | ENSE00001441774 | 0.0 | Nontriplex | ||
| exon | ENSE00002033127 | 0.0 | Nontriplex | ||
| exon | ENSE00002081221 | 0.0 | Nontriplex | ||
| exon | ENSE00002046518 | 0.0 | Nontriplex | ||
| intron 1 | 0.0 | Nontriplex | |||
| intron 2 | 0.0 | Nontriplex | |||
| intron 3 | 0.0 | Nontriplex | |||
| intron 4 | 0.124810636 | Nontriplex | |||
| intron 5 | 0.0 | Nontriplex | |||
| LINC00939 | ENST00000545784.1 | exon | ENSE00002206498 | 0.0 | Nontriplex |
| exon | ENSE00002044944 | 0.0 | Nontriplex | ||
| exon | ENSE00002228181 | 0.0 | Nontriplex | ||
| exon | ENSE00002271284 | 0.0 | Nontriplex | ||
| exon | ENSE00002021220 | 0.30899337 | Nontriplex | ||
| exon | ENSE00002216415 | 0.0 | Nontriplex | ||
| intron 1 | 0.0 | Nontriplex | |||
| intron 2 | 0.0 | Nontriplex | |||
| intron 3 | 0.124810636 | Nontriplex | |||
| intron 4 | 0.0 | Nontriplex | |||
| intron 5 | 0.0 | Nontriplex | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, R.S.; Assis, R.I.F.; Racca, F.; Bontempi, A.C.; da Silva, R.A.; Wiench, M.; Andia, D.C. Analyzes In Silico Indicate the lncRNAs MIR31HG and LINC00939 as Possible Epigenetic Inhibitors of the Osteogenic Differentiation in PDLCs. Genes 2023, 14, 1649. https://doi.org/10.3390/genes14081649
Ferreira RS, Assis RIF, Racca F, Bontempi AC, da Silva RA, Wiench M, Andia DC. Analyzes In Silico Indicate the lncRNAs MIR31HG and LINC00939 as Possible Epigenetic Inhibitors of the Osteogenic Differentiation in PDLCs. Genes. 2023; 14(8):1649. https://doi.org/10.3390/genes14081649
Chicago/Turabian StyleFerreira, Rogério S., Rahyza I. F. Assis, Francesca Racca, Ana Carolina Bontempi, Rodrigo A. da Silva, Malgorzata Wiench, and Denise C. Andia. 2023. "Analyzes In Silico Indicate the lncRNAs MIR31HG and LINC00939 as Possible Epigenetic Inhibitors of the Osteogenic Differentiation in PDLCs" Genes 14, no. 8: 1649. https://doi.org/10.3390/genes14081649
APA StyleFerreira, R. S., Assis, R. I. F., Racca, F., Bontempi, A. C., da Silva, R. A., Wiench, M., & Andia, D. C. (2023). Analyzes In Silico Indicate the lncRNAs MIR31HG and LINC00939 as Possible Epigenetic Inhibitors of the Osteogenic Differentiation in PDLCs. Genes, 14(8), 1649. https://doi.org/10.3390/genes14081649