
Combined Single Gene Testing and Genome Sequencing as an Effective Diagnostic Approach for Anophthalmia and Microphthalmia Patients

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Methods
2.1. Study Cohort and Clinical Examination
2.2. DNA Sequencing
2.3. Variant Prioritization
2.4. In Vitro Minigene Splice Assay
3. Results
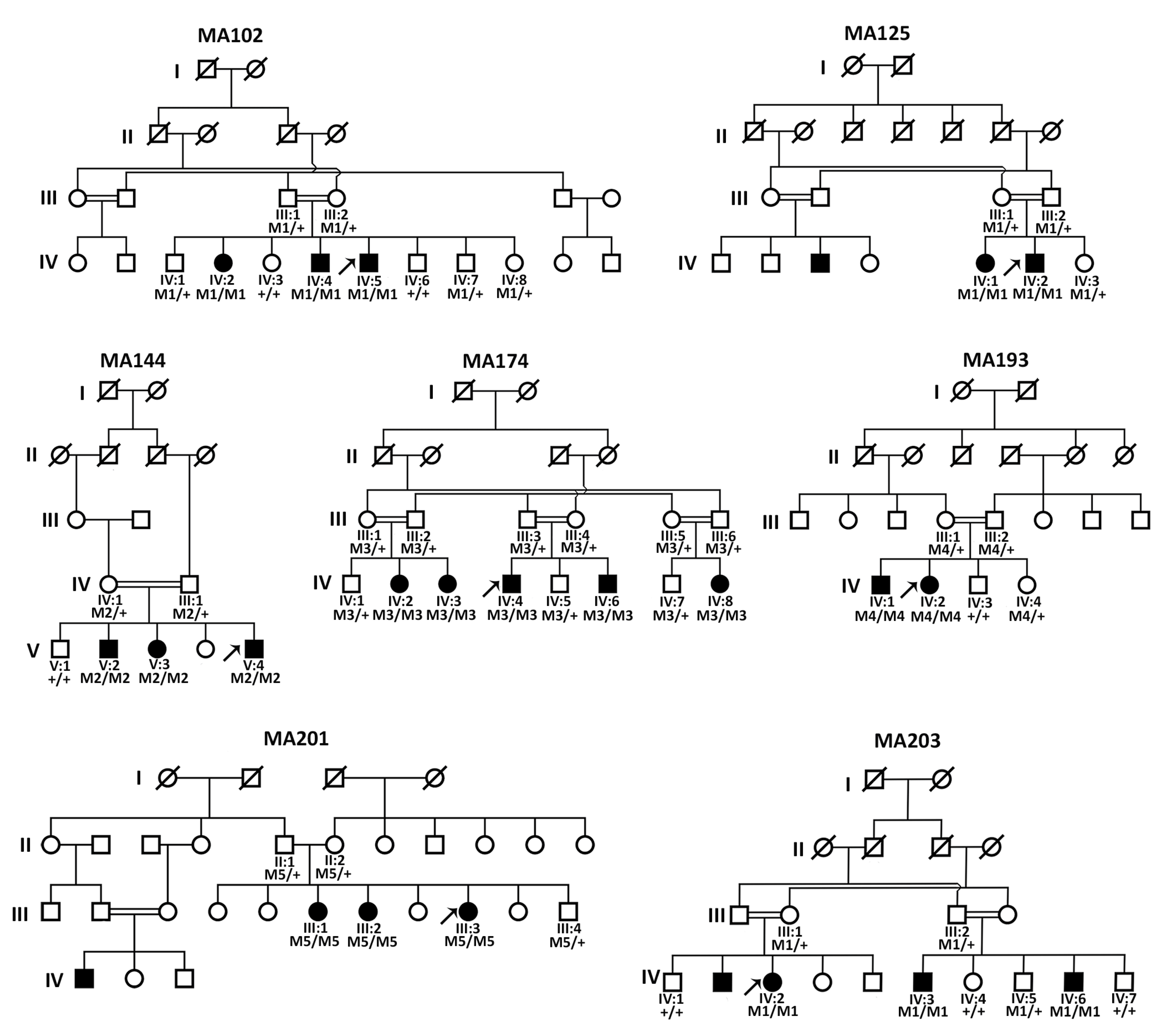
3.1. Clinical Evaluation of All Affected Members
3.2. Sanger Sequencing Revealed FOXE3 Pathogenic Variants in Four Families
3.3. Genome Sequencing Revealed Pathogenic Variants in PXDN and VSX2
3.4. Pseudoexon Activation in PXDN Caused by a Deep Intronic Splice Variant
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mann, I. The Developmental Basis of Eye Malformations; J.B. Lippincott: Philadelphia, PA, USA, 1953. [Google Scholar]
- Verma, A.S.; Fitzpatrick, D.R. Anophthalmia and microphthalmia. Orphanet J. Rare Dis. 2007, 2, 47. [Google Scholar] [CrossRef] [PubMed]
- Reis, L.M.; Semina, E.V. Genetics of anterior segment dysgenesis disorders. Curr. Opin. Ophthalmol. 2011, 22, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Searle, A.; Shetty, P.; Melov, S.J.; Alahakoon, T.I. Prenatal diagnosis and implications of microphthalmia and anophthalmia with a review of current ultrasound guidelines: Two case reports. J. Med. Case Rep. 2018, 12, 250. [Google Scholar] [CrossRef] [PubMed]
- Busby, A.; Dolk, H.; Armstrong, B. Eye anomalies: Seasonal variation and maternal viral infections. Epidemiology 2005, 16, 317–322. [Google Scholar] [CrossRef]
- Stromland, K. Visual impairment and ocular abnormalities in children with fetal alcohol syndrome. Addict. Biol. 2004, 9, 153–157, discussion 159–160. [Google Scholar] [CrossRef]
- Stromland, K.; Miller, M.T. Thalidomide embryopathy: Revisited 27 years later. Acta Ophthalmol. 1993, 71, 238–245. [Google Scholar] [CrossRef]
- Harding, P.; Moosajee, M. The Molecular Basis of Human Anophthalmia and Microphthalmia. J. Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef]
- Plaisancie, J.; Ceroni, F.; Holt, R.; Zazo Seco, C.; Calvas, P.; Chassaing, N.; Ragge, N.K. Genetics of anophthalmia and microphthalmia. Part 1: Non-syndromic anophthalmia/microphthalmia. Hum. Genet. 2019, 138, 799–830. [Google Scholar] [CrossRef]
- de Bruijn, S.E.; Rodenburg, K.; Corominas, J.; Ben-Yosef, T.; Reurink, J.; Kremer, H.; Whelan, L.; Plomp, A.S.; Berger, W.; Farrar, G.J.; et al. Optical genome mapping and revisiting short-read genome sequencing data reveal previously overlooked structural variants disrupting retinal disease-associated genes. Genet. Med. 2022, 25, 100345. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Kallberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef] [PubMed]
- Roller, E.; Ivakhno, S.; Lee, S.; Royce, T.; Tanner, S. Canvas: Versatile and scalable detection of copy number variants. Bioinformatics 2016, 32, 2375–2377. [Google Scholar] [CrossRef] [PubMed]
- Team, R. RStudio: Integrated Development for R; RStudio, PBC: Boston, MA, USA, 2020. [Google Scholar]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e524. [Google Scholar] [CrossRef]
- Zheng-Bradley, X.; Streeter, I.; Fairley, S.; Richardson, D.; Clarke, L.; Flicek, P.; Genomes Project, C. Alignment of 1000 Genomes Project reads to reference assembly GRCh38. Gigascience 2017, 6, gix038. [Google Scholar] [CrossRef]
- Hamosh, A.; Scott, A.F.; Amberger, J.S.; Bocchini, C.A.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005, 33, D514–D517. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Te Paske, I.; Mensenkamp, A.R.; Neveling, K.; Group, E.-G.L.-L.W.; Hoogerbrugge, N.; Ligtenberg, M.J.L.; De Voer, R.M. Noncoding Aberrations in Mismatch Repair Genes Underlie a Substantial Part of the Missing Heritability in Lynch Syndrome. Gastroenterology 2022, 163, 1691–1694.e1697. [Google Scholar] [CrossRef]
- Sangermano, R.; Bax, N.M.; Bauwens, M.; van den Born, L.I.; De Baere, E.; Garanto, A.; Collin, R.W.; Goercharn-Ramlal, A.S.; den Engelsman-van Dijk, A.H.; Rohrschneider, K.; et al. Photoreceptor Progenitor mRNA Analysis Reveals Exon Skipping Resulting from the ABCA4 c.5461-10T→C Mutation in Stargardt Disease. Ophthalmology 2016, 123, 1375–1385. [Google Scholar] [CrossRef]
- Ullah, E.; Nadeem Saqib, M.A.; Sajid, S.; Shah, N.; Zubair, M.; Khan, M.A.; Ahmed, I.; Ali, G.; Dutta, A.K.; Danda, S.; et al. Genetic analysis of consanguineous families presenting with congenital ocular defects. Exp. Eye Res. 2016, 146, 163–171. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Valleix, S.; Niel, F.; Nedelec, B.; Algros, M.P.; Schwartz, C.; Delbosc, B.; Delpech, M.; Kantelip, B. Homozygous nonsense mutation in the FOXE3 gene as a cause of congenital primary aphakia in humans. Am. J. Hum. Genet. 2006, 79, 358–364. [Google Scholar] [CrossRef]
- Khan, K.; Rudkin, A.; Parry, D.A.; Burdon, K.P.; McKibbin, M.; Logan, C.V.; Abdelhamed, Z.I.; Muecke, J.S.; Fernandez-Fuentes, N.; Laurie, K.J.; et al. Homozygous mutations in PXDN cause congenital cataract, corneal opacity, and developmental glaucoma. Am. J. Hum. Genet. 2011, 89, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Ammar, T.H.A.; Ismail, S.; Mansour, O.A.A.; El-Shafey, M.M.; Doghish, A.S.; Kamal, A.M.; Abdel-Salam, G.M.H. Genetic analysis of SOX2 and VSX2 genes in 27 Egyptian families with anophthalmia and microphthalmia. Ophthalmic Genet. 2017, 38, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, D.R.; van Heyningen, V. Developmental eye disorders. Curr. Opin. Genet. Dev. 2005, 15, 348–353. [Google Scholar] [CrossRef]
- Inoue, M.; Kamachi, Y.; Matsunami, H.; Imada, K.; Uchikawa, M.; Kondoh, H. PAX6 and SOX2-dependent regulation of the Sox2 enhancer N-3 involved in embryonic visual system development. Genes. Cells 2007, 12, 1049–1061. [Google Scholar] [CrossRef]
- Stigloher, C.; Ninkovic, J.; Laplante, M.; Geling, A.; Tannhauser, B.; Topp, S.; Kikuta, H.; Becker, T.S.; Houart, C.; Bally-Cuif, L. Segregation of telencephalic and eye-field identities inside the zebrafish forebrain territory is controlled by Rx3. Development 2006, 133, 2925–2935. [Google Scholar] [CrossRef]
- Winkler, S.; Loosli, F.; Henrich, T.; Wakamatsu, Y.; Wittbrodt, J. The conditional medaka mutation eyeless uncouples patterning and morphogenesis of the eye. Development 2000, 127, 1911–1919. [Google Scholar] [CrossRef]
- Reis, L.M.; Tyler, R.C.; Schneider, A.; Bardakjian, T.; Stoler, J.M.; Melancon, S.B.; Semina, E.V. FOXE3 plays a significant role in autosomal recessive microphthalmia. Am. J. Med. Genet. A 2010, 152A, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Buentello-Volante, B.; McKibbin, M.; Rocha-Medina, J.A.; Fernandez-Fuentes, N.; Koga-Nakamura, W.; Ashiq, A.; Khan, K.; Booth, A.P.; Williams, G.; et al. Homozygous FOXE3 mutations cause non-syndromic, bilateral, total sclerocornea, aphakia, microphthalmia and optic disc coloboma. Mol. Vis. 2010, 16, 1162–1168. [Google Scholar] [PubMed]
- Anjum, I.; Eiberg, H.; Baig, S.M.; Tommerup, N.; Hansen, L. A mutation in the FOXE3 gene causes congenital primary aphakia in an autosomal recessive consanguineous Pakistani family. Mol. Vis. 2010, 16, 549–555. [Google Scholar] [PubMed]
- Rashid, M.; Qasim, M.; Ishaq, R.; Bukhari, S.A.; Sajid, Z.; Ashfaq, U.A.; Haque, A.; Ahmed, Z.M. Pathogenic variants of AIPL1, MERTK, GUCY2D, and FOXE3 in Pakistani families with clinically heterogeneous eye diseases. PLoS ONE 2020, 15, e0239748. [Google Scholar] [CrossRef]
- Gillespie, R.L.; O’Sullivan, J.; Ashworth, J.; Bhaskar, S.; Williams, S.; Biswas, S.; Kehdi, E.; Ramsden, S.C.; Clayton-Smith, J.; Black, G.C.; et al. Personalized diagnosis and management of congenital cataract by next-generation sequencing. Ophthalmology 2014, 121, 2124–2137.e2. [Google Scholar] [CrossRef]
- Liu, I.S.; Chen, J.D.; Ploder, L.; Vidgen, D.; van der Kooy, D.; Kalnins, V.I.; McInnes, R.R. Developmental expression of a novel murine homeobox gene (Chx10): Evidence for roles in determination of the neuroretina and inner nuclear layer. Neuron 1994, 13, 377–393. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Khan, A.O.; Aldahmesh, M.A.; Noor, J.; Salem, A.; Alkuraya, F.S. Lens subluxation and retinal dysfunction in a girl with homozygous VSX2 mutation. Ophthalmic Genet. 2015, 36, 8–13. [Google Scholar] [CrossRef]
- Zazo-Seco, C.; Plaisancie, J.; Bitoun, P.; Corton, M.; Arteche, A.; Ayuso, C.; Schneider, A.; Zafeiropoulou, D.; Gilissen, C.; Roche, O.; et al. Novel PXDN biallelic variants in patients with microphthalmia and anterior segment dysgenesis. J. Hum. Genet. 2020, 65, 487–491. [Google Scholar] [CrossRef]
- Zhu, A.Y.; Costain, G.; Cytrynbaum, C.; Weksberg, R.; Cohn, R.D.; Ali, A. Novel heterozygous variants in PXDN cause different anterior segment dysgenesis phenotypes in monozygotic twins. Ophthalmic Genet. 2021, 42, 624–630. [Google Scholar] [CrossRef]
- Abouzeid, H.; Youssef, M.A.; Bayoumi, N.; ElShakankiri, N.; Marzouk, I.; Hauser, P.; Schorderet, D.F. RAX and anophthalmia in humans: Evidence of brain anomalies. Mol. Vis. 2012, 18, 1449–1456. [Google Scholar] [PubMed]
- Chassaing, N.; Ragge, N.; Plaisancie, J.; Patat, O.; Genevieve, D.; Rivier, F.; Malrieu-Eliaou, C.; Hamel, C.; Kaplan, J.; Calvas, P. Confirmation of TENM3 involvement in autosomal recessive colobomatous microphthalmia. Am. J. Med. Genet. A 2016, 170, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Esmailpour, T.; Riazifar, H.; Liu, L.; Donkervoort, S.; Huang, V.H.; Madaan, S.; Shoucri, B.M.; Busch, A.; Wu, J.; Towbin, A.; et al. A splice donor mutation in NAA10 results in the dysregulation of the retinoic acid signalling pathway and causes Lenz microphthalmia syndrome. J. Med. Genet. 2014, 51, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Harlalka, G.V.; Hameed, A.; Reham, H.M.; Yasin, M.; Muhammad, N.; Khan, S.; Baple, E.L.; Crosby, A.H.; Saleha, S. Novel mutations in ALDH1A3 associated with autosomal recessive anophthalmia/microphthalmia, and review of the literature. BMC Med. Genet. 2018, 19, 160. [Google Scholar] [CrossRef]
- Meienberg, J.; Bruggmann, R.; Oexle, K.; Matyas, G. Clinical sequencing: Is WGS the better WES? Hum. Genet. 2016, 135, 359–362. [Google Scholar] [CrossRef]
- Harding, P.; Gore, S.; Malka, S.; Rajkumar, J.; Oluonye, N.; Moosajee, M. Real-world clinical and molecular management of 50 prospective patients with microphthalmia, anophthalmia and/or ocular coloboma. Br. J. Ophthalmol. 2022. [Google Scholar] [CrossRef]
- Jackson, D.; Malka, S.; Harding, P.; Palma, J.; Dunbar, H.; Moosajee, M. Molecular diagnostic challenges for non-retinal developmental eye disorders in the United Kingdom. Am. J. Med. Genet. C Semin. Med. Genet. 2020, 184, 578–589. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Feature | Family ID | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MA102 | MA125 | MA144 | MA174 | MA193 | MA201 | MA203 | ||||||||||||||
| IV:2 | IV:4 | IV:5 | IV:1 | IV:2 | V:2 | V:3 | V:4 | IV:2 | IV:3 | IV:4 | IV:6 | IV:8 | IV:1 | IV:2 | III:1 | III:2 | III:3 | IV:2 | IV:3 | |
| Phenotype | A | A | A | M | M | M | M | M | A | A | A | A | A | A | A | M | M | M | M | M |
| ASD | NA | NA | NA | - | + | - | - | + | NA | NA | NA | NA | NA | NA | NA | - | - | - | + | + |
| Visual acuity | NA | NA | NA | PL | PL | NLP | PL | PL | NA | NA | NA | NA | NA | NA | NA | NLP | NLP | PL | PL | PL |
| Corneal opacity | NA | NA | NA | + | + | + | RE | + | NA | NA | NA | NA | NA | NA | NA | + | + | + | + | + |
| Flat nasal bridge | + | + | + | - | - | + | + | + | + | + | + | + | + | + | + | + | + | + | - | - |
| Family ID | Gene | cDNA | Protein | gnomAD AF Total | gnomAD AF South Asian | CADD_PHRED | REVEL | SpliceAI | ACMG/AMP | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| MA102, MA125, MA203 | FOXE3 | c.720C>A | p.(Cys240*) | 0.00001334 (Hom:0, Het:2) | 0.0004148 (Hom:0, Het:2) | 36 | NA | NA | Pathogenic | Valleix et al., 2006 [26] |
| MA201 | FOXE3 | c.289A>G | p.(Ile97Val) | 0.000006695 (Hom:0, Het:1) | 0.0002126 (Hom:0, Het:1) | 24.9 | 0.77 | NA | Pathogenic | Ullah et al., 2016 [24] |
| MA144 | PXDN | c.3609-1307G>A | p.Arg1203Serfs76* | 0.000006571 (Hom:0, Het:1) | - | NA | NA | 0.97 (AG) | Likely pathogenic | This study |
| MA174 | VSX2 | c.413_425del | p.(Ser138*) | - | - | NA | NA | NA | Pathogenic | This study |
| MA193 | PXDN | c.2568del | p.(Cys857Alafs*5) | - | - | NA | NA | NA | Pathogenic | Khan et al., 2011 [27] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basharat, R.; Rodenburg, K.; Rodríguez-Hidalgo, M.; Jarral, A.; Ullah, E.; Corominas, J.; Gilissen, C.; Zehra, S.T.; Hameed, U.; Ansar, M.; et al. Combined Single Gene Testing and Genome Sequencing as an Effective Diagnostic Approach for Anophthalmia and Microphthalmia Patients. Genes 2023, 14, 1573. https://doi.org/10.3390/genes14081573
Basharat R, Rodenburg K, Rodríguez-Hidalgo M, Jarral A, Ullah E, Corominas J, Gilissen C, Zehra ST, Hameed U, Ansar M, et al. Combined Single Gene Testing and Genome Sequencing as an Effective Diagnostic Approach for Anophthalmia and Microphthalmia Patients. Genes. 2023; 14(8):1573. https://doi.org/10.3390/genes14081573
Chicago/Turabian StyleBasharat, Rabia, Kim Rodenburg, María Rodríguez-Hidalgo, Afeefa Jarral, Ehsan Ullah, Jordi Corominas, Christian Gilissen, Syeda Tatheer Zehra, Usman Hameed, Muhammad Ansar, and et al. 2023. "Combined Single Gene Testing and Genome Sequencing as an Effective Diagnostic Approach for Anophthalmia and Microphthalmia Patients" Genes 14, no. 8: 1573. https://doi.org/10.3390/genes14081573
APA StyleBasharat, R., Rodenburg, K., Rodríguez-Hidalgo, M., Jarral, A., Ullah, E., Corominas, J., Gilissen, C., Zehra, S. T., Hameed, U., Ansar, M., & de Bruijn, S. E. (2023). Combined Single Gene Testing and Genome Sequencing as an Effective Diagnostic Approach for Anophthalmia and Microphthalmia Patients. Genes, 14(8), 1573. https://doi.org/10.3390/genes14081573