
The DEAD-Box RNA Helicase Ded1 Is Associated with Translating Ribosomes


Abstract
1. Introduction
2. Materials and Methods
2.1. Constructs, Yeast Strains, and Manipulations
2.2. Growth Curves
2.3. 4-thioU Incorporation and Crosslinking Conditions
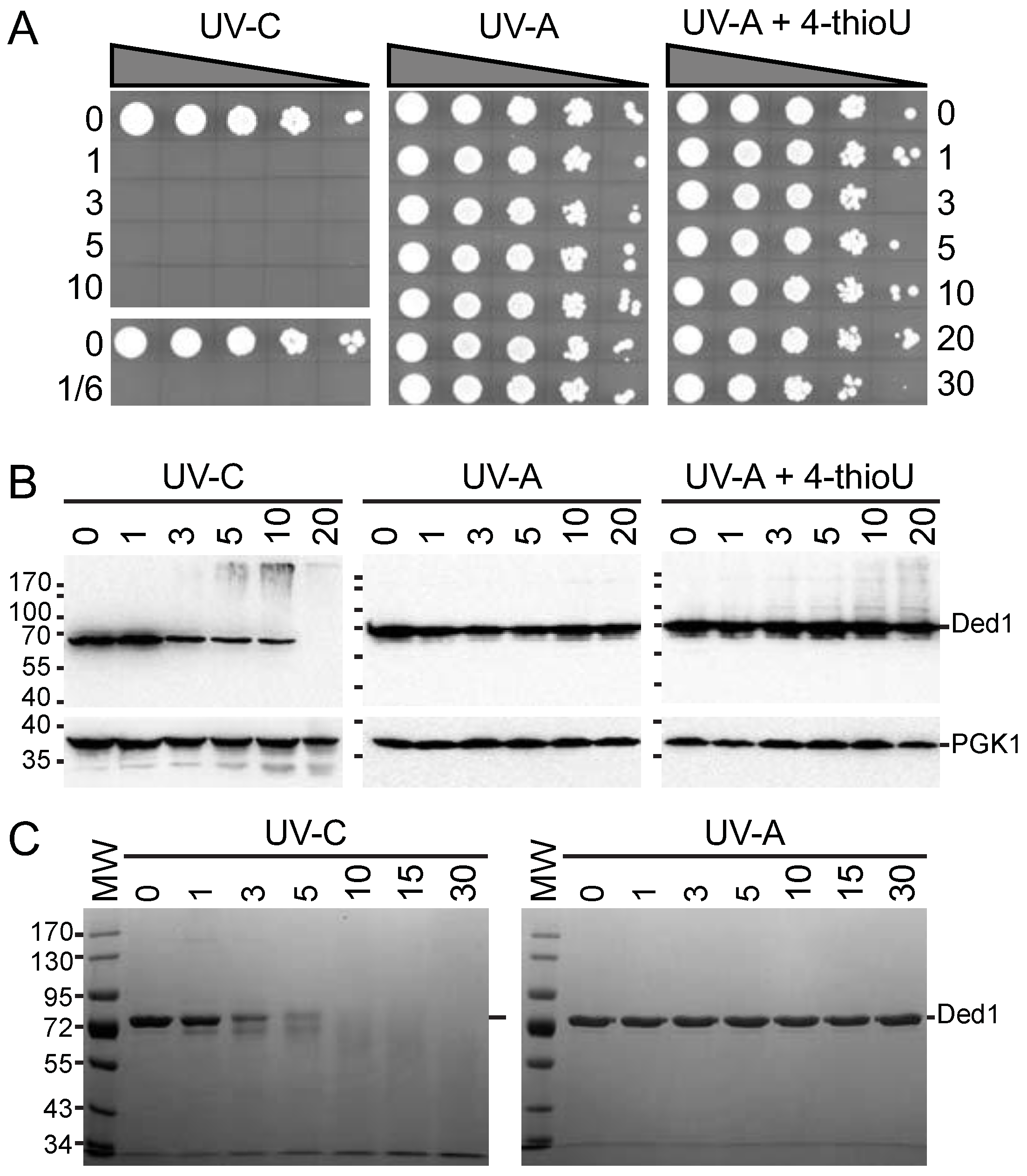
2.4. Growth Viability and Protein Stability to UV Light
2.5. qtPAR-CLIP Protocol
2.6. Processing and Analysis of Sequencing Data
2.7. Differential Expression
2.8. Polysome Analyses
2.9. Fluorescence Microscopy
3. Results
3.1. Development of the qtPAR-CLIP Technique
3.1.1. The 4-thioU and UV-A Light Yield Crosslinks under Physiological Conditions
3.1.2. Optimization of the qtPAR-CLIP Conditions
3.2. Ded1 Crosslinks to Specific RNAs Often at Specific Sites
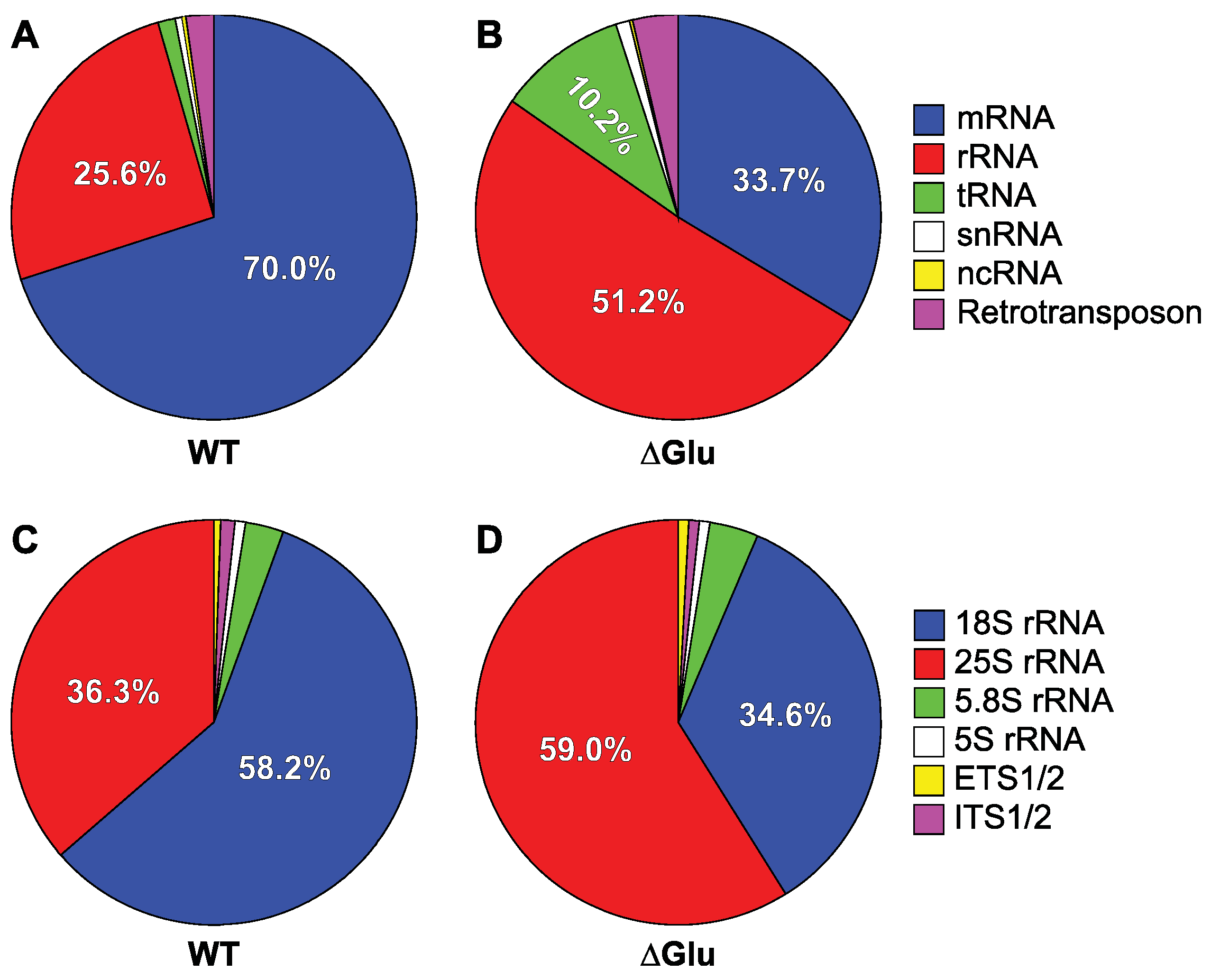
3.2.1. Ded1 Crosslinks Mostly to mRNAs
3.2.2. Glucose Depletion Redistributes the Crosslinks on rRNAs
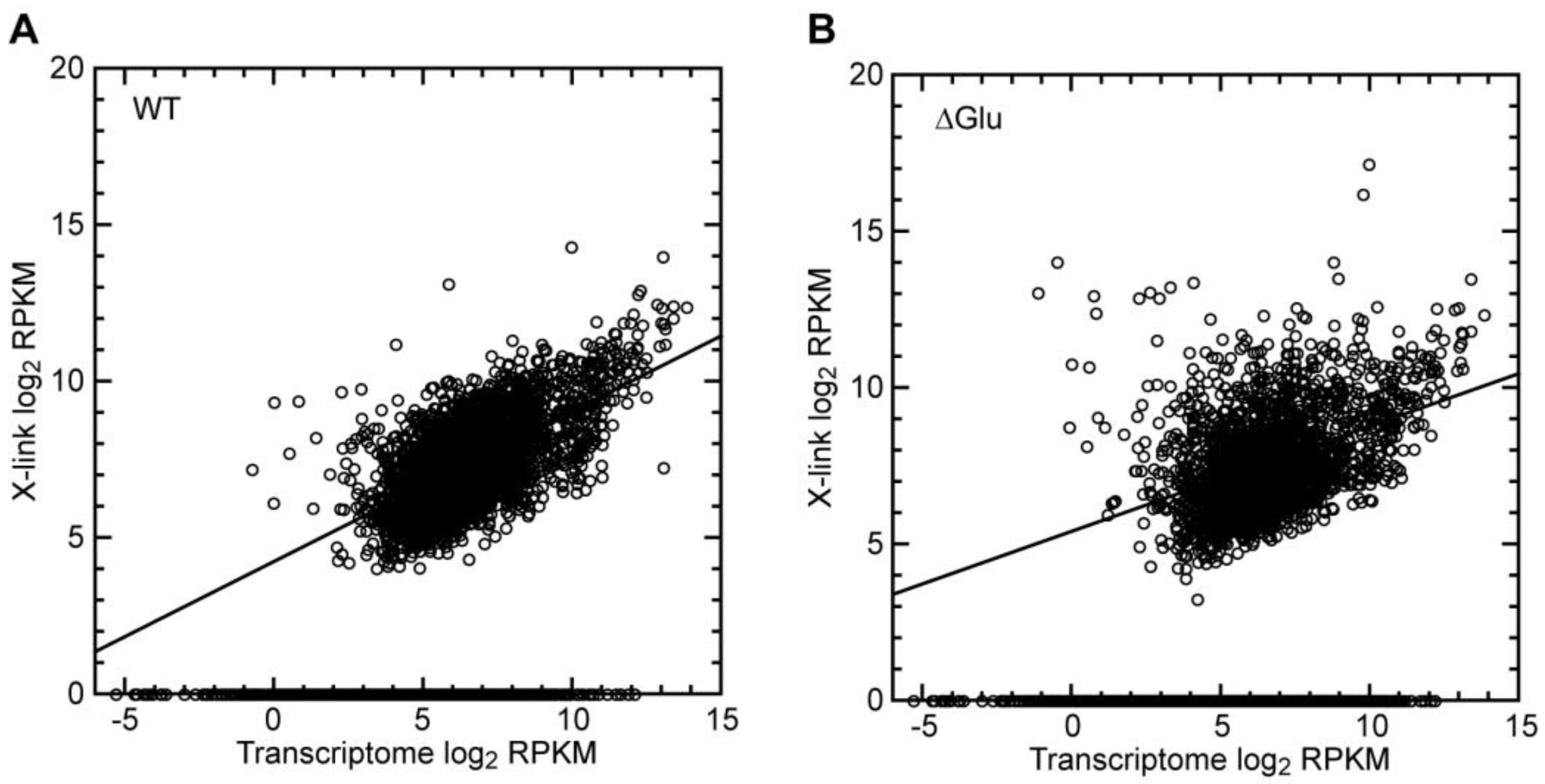
3.2.3. Crosslinks Partially Correlated with mRNA Abundance
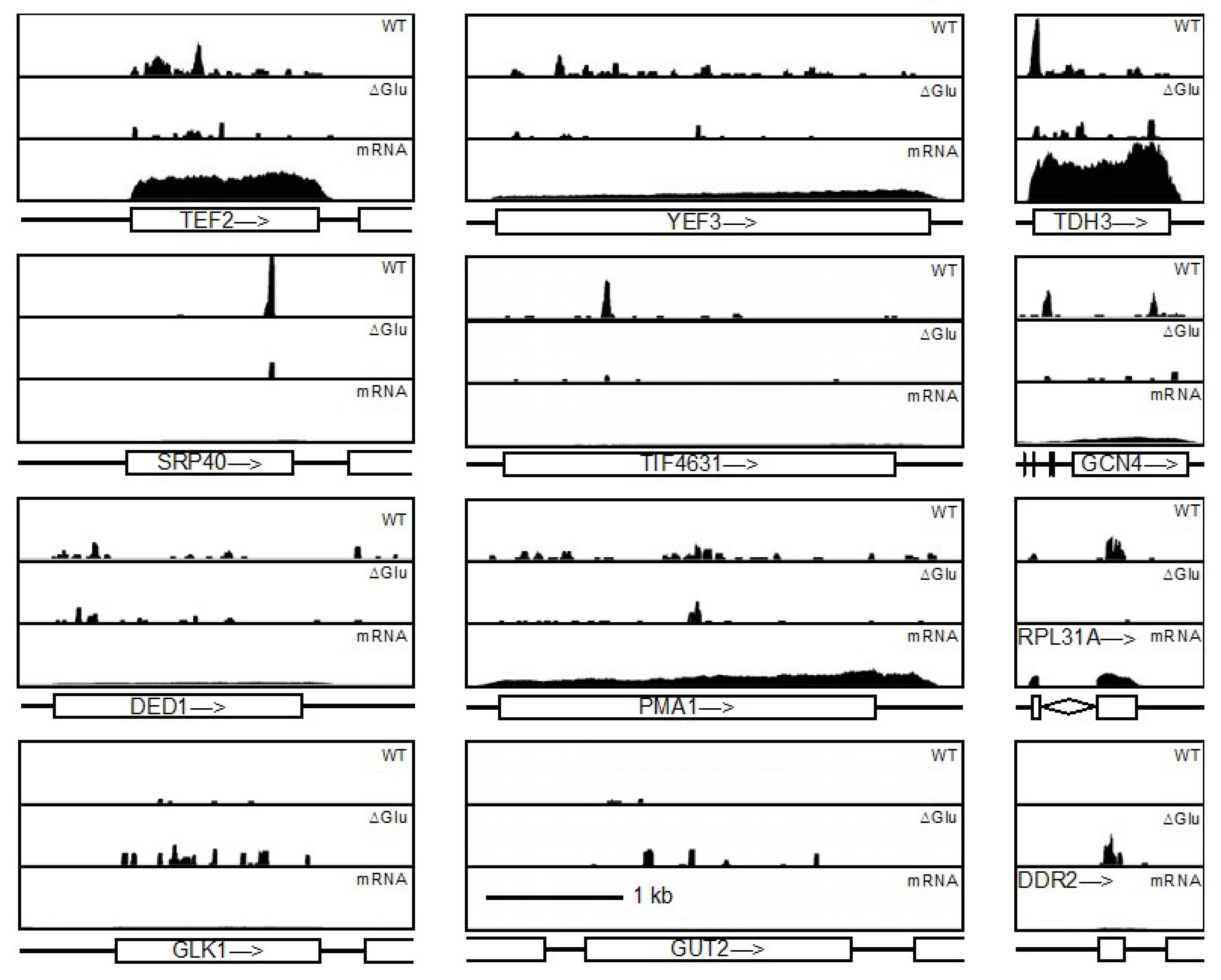
3.2.4. Ded1 Was Highly Crosslinked to Few mRNAs
3.2.5. Glucose-Depletion Crosslinks Reflected Altered Genetic Expression
3.2.6. Crosslinks Poorly Correlated with mRNAs UTR Length
3.2.7. Ded1 Preferentially Binds Purine-Rich Sequences
3.2.8. Ded1 Crosslinked to tRNAs
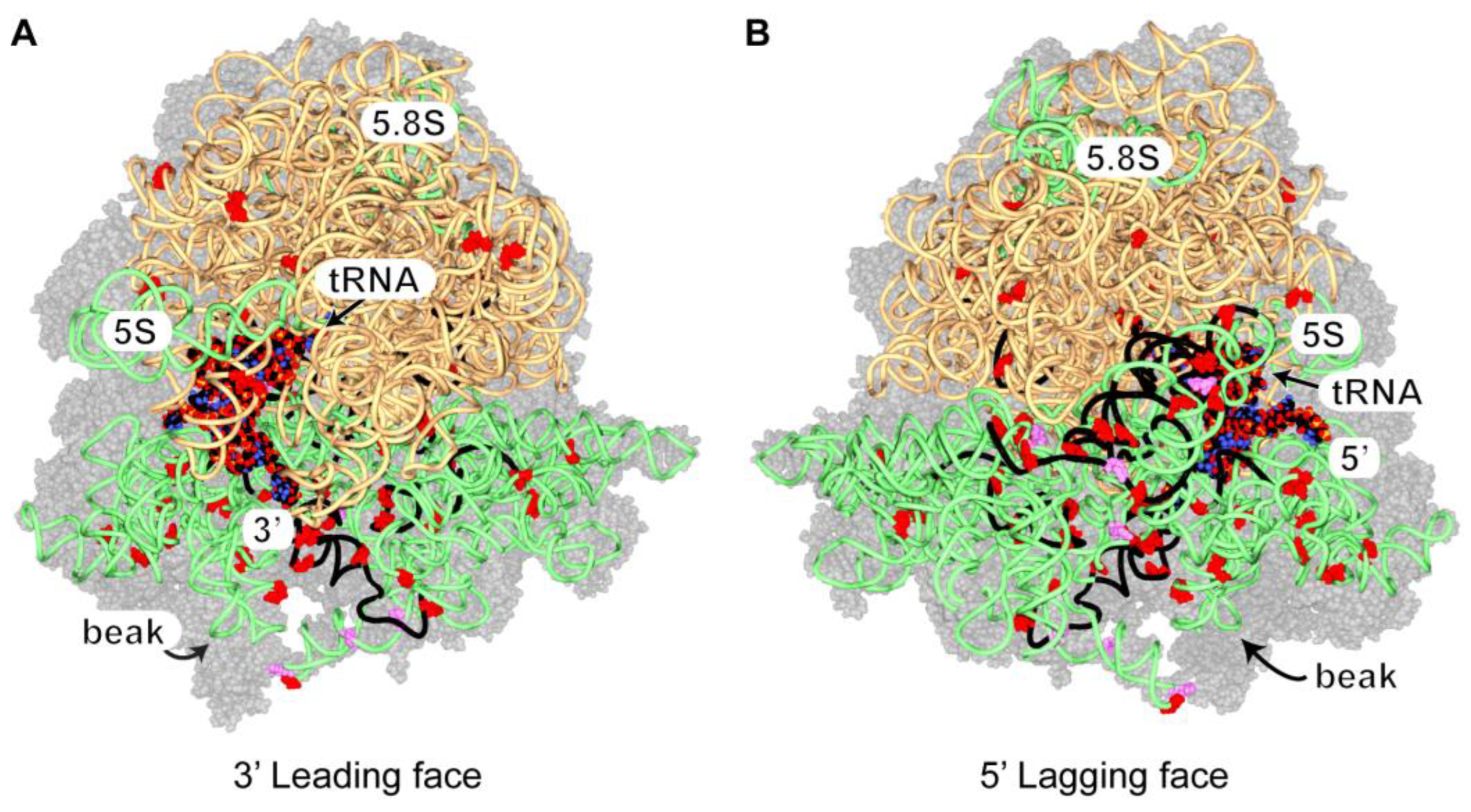
3.2.9. Ded1 Crosslinks on 80S Ribosomes

3.3. A Subpopulation of Ded1 Was in Close Proximity to the ER
3.4. Ded1 Was Associated Transiently with Polysomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cargill, M.; Venkataraman, R.; Lee, S. DEAD-Box RNA Helicases and Genome Stability. Genes 2021, 12, 1471. [Google Scholar] [CrossRef] [PubMed]
- Cordin, O.; Banroques, J.; Tanner, N.K.; Linder, P. The DEAD-box protein family of RNA helicases. Gene 2006, 367, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Linder, P.; Jankowsky, E. From unwinding to clamping—The DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011, 12, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Putnam, A.A.; Jankowsky, E. DEAD-box helicases as integrators of RNA, nucleotide and protein binding. Biochim. Biophys. Acta 2013, 1829, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Weis, K.; Hondele, M. The Role of DEAD-Box ATPases in Gene Expression and the Regulation of RNA-Protein Condensates. Annu. Rev. Biochem. 2022, 91, 197–219. [Google Scholar] [CrossRef] [PubMed]
- Ozgur, S.; Buchwald, G.; Falk, S.; Chakrabarti, S.; Prabu, J.R.; Conti, E. The conformational plasticity of eukaryotic RNA-dependent ATPases. FEBS J. 2015, 282, 850–863. [Google Scholar] [CrossRef]
- Schütz, P.; Karlberg, T.; van den Berg, S.; Collins, R.; Lehtiö, L.; Högbom, M.; Holmberg-Schiavone, L.; Tempel, W.; Park, H.W.; Hammarström, M.; et al. Comparative structural analysis of human DEAD-box RNA helicases. PLoS ONE 2010, 5, e12791. [Google Scholar] [CrossRef]
- Liu, F.; Putnam, A.A.; Jankowsky, E. DEAD-box helicases form nucleotide-dependent, long-lived complexes with RNA. Biochemistry 2014, 53, 423–433. [Google Scholar] [CrossRef]
- Tanner, N.K.; Linder, P. DExD/H box RNA helicases: From generic motors to specific dissociation functions. Mol. Cell 2001, 8, 251–262. [Google Scholar] [CrossRef]
- de la Cruz, J.; Kressler, D.; Linder, P. Unwinding RNA in Saccharomyces cerevisiae: DEAD-box proteins and related families. Trends Biochem. Sci. 1999, 24, 192–198. [Google Scholar] [CrossRef]
- Banroques, J.; Cordin, O.; Doère, M.; Linder, P.; Tanner, N.K. Analyses of the functional regions of DEAD-box RNA “helicases” with deletion and chimera constructs tested in vivo and in vitro. J. Mol. Biol. 2011, 413, 451–472. [Google Scholar] [CrossRef] [PubMed]
- Fairman-Williams, M.E.; Guenther, U.P.; Jankowsky, E. SF1 and SF2 helicases: Family matters. Curr. Opin. Struct. Biol. 2010, 20, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Umate, P.; Tuteja, N.; Tuteja, R. Genome-wide comprehensive analysis of human helicases. Commun. Integr. Biol. 2011, 4, 118–137. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.C.; Liu, W.S. The molecular evolution of PL10 homologs. BMC Evol. Biol. 2010, 10, 127. [Google Scholar] [CrossRef]
- Rosner, A.; Rinkevich, B. The DDX3 subfamily of the DEAD box helicases: Divergent roles as unveiled by studying different organisms and in vitro assays. Curr. Med. Chem. 2007, 14, 2517–2525. [Google Scholar] [CrossRef]
- Ryan, C.S.; Schröder, M. The human DEAD-box helicase DDX3X as a regulator of mRNA translation. Front. Cell Dev. Biol. 2022, 10, 1033684. [Google Scholar] [CrossRef]
- Sharma, D.; Jankowsky, E. The Ded1/DDX3 subfamily of DEAD-box RNA helicases. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 343–360. [Google Scholar] [CrossRef]
- Tarn, W.Y.; Chang, T.H. The current understanding of Ded1p/DDX3 homologs from yeast to human. RNA Biol. 2009, 6, 17–20. [Google Scholar] [CrossRef]
- Senissar, M.; Le Saux, A.; Belgareh-Touzé, N.; Adam, C.; Banroques, J.; Tanner, N.K. The DEAD-box helicase Ded1 from yeast is an mRNP cap-associated protein that shuttles between the cytoplasm and nucleus. Nucleic Acids Res. 2014, 42, 10005–10022. [Google Scholar] [CrossRef]
- Jamieson, D.J.; Rahe, B.; Pringle, J.; Beggs, J.D. A suppressor of a yeast splicing mutation (prp8-1) encodes a putative ATP-dependent RNA helicase. Nature 1991, 349, 715–717. [Google Scholar] [CrossRef][Green Version]
- Thuillier, V.; Stettler, S.; Sentenac, A.; Thuriaux, P.; Werner, M. A mutation in the C31 subunit of Saccharomyces cerevisiae RNA polymerase III affects transcription initiation. EMBO J. 1995, 14, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Chuang, R.Y.; Weaver, P.L.; Liu, Z.; Chang, T.H. Requirement of the DEAD-Box protein ded1p for messenger RNA translation. Science 1997, 275, 1468–1471. [Google Scholar] [CrossRef] [PubMed]
- de la Cruz, J.; Iost, I.; Kressler, D.; Linder, P. The p20 and Ded1 proteins have antagonistic roles in eIF4E-dependent translation in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 5201–5206. [Google Scholar] [CrossRef]
- Berthelot, K.; Muldoon, M.; Rajkowitsch, L.; Hughes, J.; McCarthy, J.E. Dynamics and processivity of 40S ribosome scanning on mRNA in yeast. Mol. Microbiol. 2004, 51, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.L.; Chuang, R.Y.; Tung, L.; Chang, T.H. Ded1p, a conserved DExD/H-box translation factor, can promote yeast L-A virus negative-strand RNA synthesis in vitro. Nucleic Acids Res. 2004, 32, 2031–2038. [Google Scholar] [CrossRef]
- Aryanpur, P.P.; Renner, D.M.; Rodela, E.; Mittelmeier, T.M.; Byrd, A.; Bolger, T.A. The DEAD-box RNA helicase Ded1 has a role in the translational response to TORC1 inhibition. Mol. Biol. Cell 2019, 30, 2171–2184. [Google Scholar] [CrossRef]
- Aryanpur, P.P.; Mittelmeier, T.M.; Bolger, T.A. The RNA Helicase Ded1 Regulates Translation and Granule Formation during Multiple Phases of Cellular Stress Responses. Mol. Cell Biol. 2022, 42, e0024421. [Google Scholar] [CrossRef]
- Beckham, C.; Hilliker, A.; Cziko, A.M.; Noueiry, A.; Ramaswami, M.; Parker, R. The DEAD-box RNA helicase Ded1p affects and accumulates in Saccharomyces cerevisiae P-bodies. Mol. Biol. Cell 2008, 19, 984–993. [Google Scholar] [CrossRef]
- Guzikowski, A.R.; Chen, Y.S.; Zid, B.M. Stress-induced mRNP granules: Form and function of processing bodies and stress granules. Wiley Interdiscip. Rev. RNA 2019, 10, e1524. [Google Scholar] [CrossRef]
- Hondele, M.; Sachdev, R.; Heinrich, S.; Wang, J.; Vallotton, P.; Fontoura, B.M.A.; Weis, K. DEAD-box ATPases are global regulators of phase-separated organelles. Nature 2019, 573, 144–148. [Google Scholar] [CrossRef]
- Ivanov, P.; Kedersha, N.; Anderson, P. Stress Granules and Processing Bodies in Translational Control. Cold Spring Harb. Perspect. Biol. 2019, 11, a032813. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Díaz, T.; Valiente-Echeverría, F.; Soto-Rifo, R. RNA Helicase DDX3: A Double-Edged Sword for Viral Replication and Immune Signaling. Microorganisms 2021, 9, 1206. [Google Scholar] [CrossRef] [PubMed]
- Soto-Rifo, R.; Ohlmann, T. The role of the DEAD-box RNA helicase DDX3 in mRNA metabolism. Wiley Interdiscip. Rev. RNA 2013, 4, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Calviello, L.; Venkataramanan, S.; Rogowski, K.J.; Wyler, E.; Wilkins, K.; Tejura, M.; Thai, B.; Krol, J.; Filipowicz, W.; Landthaler, M.; et al. DDX3 depletion represses translation of mRNAs with complex 5′ UTRs. Nucleic Acids Res. 2021, 49, 5336–5350. [Google Scholar] [CrossRef]
- Lai, M.C.; Chang, W.C.; Shieh, S.Y.; Tarn, W.Y. DDX3 regulates cell growth through translational control of cyclin E1. Mol. Cell. Biol. 2010, 30, 5444–5453. [Google Scholar] [CrossRef]
- Padmanabhan, P.K.; Ferreira, G.R.; Zghidi-Abouzid, O.; Oliveira, C.; Dumas, C.; Mariz, F.C.; Papadopoulou, B. Genetic depletion of the RNA helicase DDX3 leads to impaired elongation of translating ribosomes triggering co-translational quality control of newly synthesized polypeptides. Nucleic Acids Res. 2021, 49, 9459–9478. [Google Scholar] [CrossRef]
- Zhao, L.; Mao, Y.; Zhou, J.; Zhao, Y.; Cao, Y.; Chen, X. Multifunctional DDX3: Dual roles in various cancer development and its related signaling pathways. Am. J. Cancer Res. 2016, 6, 387–402. [Google Scholar]
- Hauk, G.; Bowman, G.D. Formation of a Trimeric Xpo1-Ran[GTP]-Ded1 Exportin Complex Modulates ATPase and Helicase Activities of Ded1. PLoS ONE 2015, 10, e0131690. [Google Scholar] [CrossRef]
- Fröhlich, A.; Rojas-Araya, B.; Pereira-Montecinos, C.; Dellarossa, A.; Toro-Ascuy, D.; Prades-Pérez, Y.; García-de-Gracia, F.; Garcés-Alday, A.; Rubilar, P.S.; Valiente-Echeverría, F.; et al. DEAD-box RNA helicase DDX3 connects CRM1-dependent nuclear export and translation of the HIV-1 unspliced mRNA through its N-terminal domain. Biochim. Biophys. Acta 2016, 1859, 719–730. [Google Scholar] [CrossRef]
- Bresson, S.; Shchepachev, V.; Spanos, C.; Turowski, T.W.; Rappsilber, J.; Tollervey, D. Stress-Induced Translation Inhibition through Rapid Displacement of Scanning Initiation Factors. Mol. Cell 2020, 80, 470–484.e478. [Google Scholar] [CrossRef]
- Guenther, U.P.; Weinberg, D.E.; Zubradt, M.M.; Tedeschi, F.A.; Stawicki, B.N.; Zagore, L.L.; Brar, G.A.; Licatalosi, D.D.; Bartel, D.P.; Weissman, J.S.; et al. The helicase Ded1p controls use of near-cognate translation initiation codons in 5′ UTRs. Nature 2018, 559, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Gulay, S.; Gupta, N.; Lorsch, J.R.; Hinnebusch, A.G. Distinct interactions of eIF4A and eIF4E with RNA helicase Ded1 stimulate translation in vivo. eLife 2020, 9, 58243. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Lorsch, J.R.; Hinnebusch, A.G. Yeast Ded1 promotes 48S translation pre-initiation complex assembly in an mRNA-specific and eIF4F-dependent manner. eLife 2018, 7, 38892. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.D.; Gupta, N.; Archer, S.K.; Preiss, T.; Lorsch, J.R.; Hinnebusch, A.G. Functional interplay between DEAD-box RNA helicases Ded1 and Dbp1 in preinitiation complex attachment and scanning on structured mRNAs in vivo. Nucleic Acids Res. 2019, 47, 8785–8806. [Google Scholar] [CrossRef]
- Brito Querido, J.; Sokabe, M.; Kraatz, S.; Gordiyenko, Y.; Skehel, J.M.; Fraser, C.S.; Ramakrishnan, V. Structure of a human 48S translational initiation complex. Science 2020, 369, 1220–1227. [Google Scholar] [CrossRef]
- Hinnebusch, A.G. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev. MMBR 2011, 75, 434–467. [Google Scholar] [CrossRef]
- Spirin, A.S. How Does a Scanning Ribosomal Particle Move along the 5′-Untranslated Region of Eukaryotic mRNA? Brownian Ratchet Model. Biochemistry 2009, 48, 10688–10692. [Google Scholar] [CrossRef]
- Sen, N.D.; Zhang, H.; Hinnebusch, A.G. Down-Regulation of Yeast Helicase Ded1 by Glucose Starvation or Heat-Shock Differentially Impairs Translation of Ded1-Dependent mRNAs. Microorganisms 2021, 9, 2413. [Google Scholar] [CrossRef]
- Attri, P.; Kim, Y.H.; Park, D.H.; Park, J.H.; Hong, Y.J.; Uhm, H.S.; Kim, K.N.; Fridman, A.; Choi, E.H. Generation mechanism of hydroxyl radical species and its lifetime prediction during the plasma-initiated ultraviolet (UV) photolysis. Sci. Rep. 2015, 5, 9332. [Google Scholar] [CrossRef]
- Hafner, M.; Katsantoni, M.; Köster, T.; Marks, J.; Mukherjee, J.; Staiger, D.; Ule, J.; Zavolan, M. CLIP and complementary methods. Nat. Rev. Methods Prim. 2021, 1, 20. [Google Scholar] [CrossRef]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef]
- Meisenheimer, K.M.; Koch, T.H. Photocross-linking of nucleic acids to associated proteins. Crit. Rev. Biochem. Mol. Biol. 1997, 32, 101–140. [Google Scholar] [CrossRef]
- Tanner, N.K.; Hanna, M.M.; Abelson, J. Binding interactions between yeast tRNA ligase and a precursor transfer ribonucleic acid containing two photoreactive uridine analogues. Biochemistry 1988, 27, 8852–8861. [Google Scholar] [CrossRef] [PubMed]
- Iost, I.; Dreyfus, M.; Linder, P. Ded1p, a DEAD-box protein required for translation initiation in Saccharomyces cerevisiae, is an RNA helicase. J. Biol. Chem. 1999, 274, 17677–17683. [Google Scholar] [CrossRef]
- Deshaies, R.J.; Schekman, R. A yeast mutant defective at an early stage in import of secretory protein precursors into the endoplasmic reticulum. J. Cell Biol. 1987, 105, 633–645. [Google Scholar] [CrossRef]
- Panova, N.G.; Shcheveleva, E.V.; Alexeev, C.S.; Mukhortov, V.G.; Zuev, A.N.; Mikhailov, S.N.; Esipov, R.S.; Chuvikovsky, D.V.; Miroshnikov, A.I. Use of 4-Thiouridine and 4-Thiothymidine in Studies on Pyrimidine Nucleoside Phosphorylases. Mol. Biol. 2004, 38, 770–776. [Google Scholar] [CrossRef]
- Beckmann, B.M. RNA interactome capture in yeast. Methods 2017, 118–119, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhou, C.; Jiang, M. UV absorption spectra of polystyrene. Polym. Bull. 1991, 25, 211–216. [Google Scholar] [CrossRef]
- Horvath, A.; Riezman, H. Rapid protein extraction from Saccharomyces cerevisiae. Yeast. 1994, 10, 1305–1310. [Google Scholar] [CrossRef]
- Spitzer, J.; Hafner, M.; Landthaler, M.; Ascano, M.; Farazi, T.; Wardle, G.; Nusbaum, J.; Khorshid, M.; Burger, L.; Zavolan, M.; et al. PAR-CLIP (Photoactivatable Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation): A step-by-step protocol to the transcriptome-wide identification of binding sites of RNA-binding proteins. Methods Enzymol. 2014, 539, 113–161. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Kishore, S.; Jaskiewicz, L.; Burger, L.; Hausser, J.; Khorshid, M.; Zavolan, M. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat Methods. 2011, 8, 559–564. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Varet, H.; Brillet-Guéguen, L.; Coppée, J.Y.; Dillies, M.A. SARTools: A DESeq2- and EdgeR-Based R Pipeline for Comprehensive Differential Analysis of RNA-Seq Data. PLoS ONE 2016, 11, e0157022. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J.; Schekman, R. SEC62 encodes a putative membrane protein required for protein translocation into the yeast endoplasmic reticulum. J. Cell Biol. 1989, 109, 2653–2664. [Google Scholar] [CrossRef]
- Nishikawa, S.I.; Fewell, S.W.; Kato, Y.; Brodsky, J.L.; Endo, T. Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J. Cell Biol. 2001, 153, 1061–1070. [Google Scholar] [CrossRef]
- Li, X.; Cai, M.; Wang, L.; Niu, F.; Yang, D.; Zhang, G. Evaluation survey of microbial disinfection methods in UV-LED water treatment systems. Sci. Total Environ. 2019, 659, 1415–1427. [Google Scholar] [CrossRef] [PubMed]
- Danan, C.; Manickavel, S.; Hafner, M. PAR-CLIP: A Method for Transcriptome-Wide Identification of RNA Binding Protein Interaction Sites. Methods Mol. Biol. 2022, 2404, 167–188. [Google Scholar] [CrossRef] [PubMed]
- Zarnegar, B.J.; Flynn, R.A.; Shen, Y.; Do, B.T.; Chang, H.Y.; Khavari, P.A. irCLIP platform for efficient characterization of protein-RNA interactions. Nat. Methods 2016, 13, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.E. Zinc enzymes. Curr. Opin. Chem. Biol. 1998, 2, 222–234. [Google Scholar] [CrossRef]
- Anastasakis, D.G.; Jacob, A.; Konstantinidou, P.; Meguro, K.; Claypool, D.; Cekan, P.; Haase, A.D.; Hafner, M. A non-radioactive, improved PAR-CLIP and small RNA cDNA library preparation protocol. Nucleic Acids Res. 2021, 49, e45. [Google Scholar] [CrossRef]
- Ashe, M.P.; De Long, S.K.; Sachs, A.B. Glucose depletion rapidly inhibits translation initiation in yeast. Mol. Biol. Cell 2000, 11, 833–848. [Google Scholar] [CrossRef]
- Janapala, Y.; Preiss, T.; Shirokikh, N.E. Control of Translation at the Initiation Phase During Glucose Starvation in Yeast. Int. J. Mol. Sci. 2019, 20, 4043. [Google Scholar] [CrossRef]
- Lui, J.; Campbell, S.G.; Ashe, M.P. Inhibition of translation initiation following glucose depletion in yeast facilitates a rationalization of mRNA content. Biochem. Soc. Trans. 2010, 38, 1131–1136. [Google Scholar] [CrossRef]
- Kiss, T. Small nucleolar RNAs: An abundant group of noncoding RNAs with diverse cellular functions. Cell 2002, 109, 145–148. [Google Scholar] [CrossRef]
- Nyathi, Y.; Wilkinson, B.M.; Pool, M.R. Co-translational targeting and translocation of proteins to the endoplasmic reticulum. Biochim. Et Biophys. Acta 2013, 1833, 2392–2402. [Google Scholar] [CrossRef]
- Sokabe, M.; Fraser, C.S. Toward a Kinetic Understanding of Eukaryotic Translation. Cold Spring Harb. Perspect. Biol. 2019, 11, a032706. [Google Scholar] [CrossRef] [PubMed]
- Yeter-Alat, H.; Belgareh-Touzé, N.; Huvelle, E.; Mokdadi, M.; Banroques, J.; Tanner, N.K. The DEAD-box RNA helicase Ded1 from yeast is associated with the signal recognition particle (SRP), and its enzymatic activity is regulated by SRP21. bioRxiv 2020. [Google Scholar] [CrossRef]
- Shuman, S. Transcriptional interference at tandem lncRNA and protein-coding genes: An emerging theme in regulation of cellular nutrient homeostasis. Nucleic Acids Res. 2020, 48, 8243–8254. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.M.; Parker, R. Cytoplasmic decay of intergenic transcripts in Saccharomyces cerevisiae. Mol. Cell Biol. 2007, 27, 92–101. [Google Scholar] [CrossRef]
- van den Elzen, A.M.; Schuller, A.; Green, R.; Séraphin, B. Dom34-Hbs1 mediated dissociation of inactive 80S ribosomes promotes restart of translation after stress. EMBO J. 2014, 33, 265–276. [Google Scholar] [CrossRef]
- Crawford, R.A.; Pavitt, G.D. Translational regulation in response to stress in Saccharomyces cerevisiae. Yeast 2019, 36, 5–21. [Google Scholar] [CrossRef]
- Oh, S.; Flynn, R.A.; Floor, S.N.; Purzner, J.; Martin, L.; Do, B.T.; Schubert, S.; Vaka, D.; Morrissy, S.; Li, Y.; et al. Medulloblastoma-associated DDX3 variant selectively alters the translational response to stress. Oncotarget 2016, 7, 28169–28182. [Google Scholar] [CrossRef]
- An, H.; Harper, J.W. Ribosome Abundance Control Via the Ubiquitin-Proteasome System and Autophagy. J. Mol. Biol. 2020, 432, 170–184. [Google Scholar] [CrossRef]
- Rodríguez, A.; De La Cera, T.; Herrero, P.; Moreno, F. The hexokinase 2 protein regulates the expression of the GLK1, HXK1 and HXK2 genes of Saccharomyces cerevisiae. Biochem. J. 2001, 355, 625–631. [Google Scholar] [CrossRef]
- Lin, Z.; Li, W.H. Evolution of 5′ untranslated region length and gene expression reprogramming in yeasts. Mol. Biol. Evol. 2012, 29, 81–89. [Google Scholar] [CrossRef]
- Pelechano, V.; Wei, W.; Steinmetz, L.M. Extensive transcriptional heterogeneity revealed by isoform profiling. Nature 2013, 497, 127–131. [Google Scholar] [CrossRef]
- Mayr, C. What Are 3′ UTRs Doing? Cold Spring Harb. Perspect. Biol. 2019, 11, a034728. [Google Scholar] [CrossRef] [PubMed]
- Freese, N.H.; Norris, D.C.; Loraine, A.E. Integrated genome browser: Visual analytics platform for genomics. Bioinformatics 2016, 32, 2089–2095. [Google Scholar] [CrossRef] [PubMed]
- Lawless, C.; Pearson, R.D.; Selley, J.N.; Smirnova, J.B.; Grant, C.M.; Ashe, M.P.; Pavitt, G.D.; Hubbard, S.J. Upstream sequence elements direct post-transcriptional regulation of gene expression under stress conditions in yeast. BMC Genom. 2009, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, S.A.; Ogurtsov, A.Y.; Rogozin, I.B.; Koonin, E.V.; Lipman, D.J. Comparative analysis of orthologous eukaryotic mRNAs: Potential hidden functional signals. Nucleic Acids Res. 2004, 32, 1774–1782. [Google Scholar] [CrossRef]
- Uppala, J.K.; Sathe, L.; Chakraborty, A.; Bhattacharjee, S.; Pulvino, A.T.; Dey, M. The cap-proximal RNA secondary structure inhibits preinitiation complex formation on HAC1 mRNA. J. Biol. Chem. 2022, 298, 101648. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef]
- Wickner, R.B. Double-stranded RNA viruses of Saccharomyces cerevisiae. Microbiol. Rev. 1996, 60, 250–265. [Google Scholar] [CrossRef]
- Curcio, M.J.; Lutz, S.; Lesage, P. The Ty1 LTR-Retrotransposon of Budding Yeast, Saccharomyces cerevisiae. Microbiol. Spectr. 2015, 3, 1–35. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H. Diverse transposable element landscapes in pathogenic and nonpathogenic yeast models: The value of a comparative perspective. Mob. DNA 2020, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Doh, J.H.; Lutz, S.; Curcio, M.J. Co-translational localization of an LTR-retrotransposon RNA to the endoplasmic reticulum nucleates virus-like particle assembly sites. PLoS Genet. 2014, 10, e1004219. [Google Scholar] [CrossRef] [PubMed]
- Abdelkrim, Y.Z.; Mokdadi, M.; Banroques, J.; Huvelle, E.; Yeter-Alat, H.; Crobu, L.; Sterkers, Y.; Guizani, I.; Barhoumi, M.; Tanner, N.K. Identification and characterization of a PRP5-like DEAD-box “helicase” with unusual enzymatic properties from the Trypanosomatid parasite Leishmania infantum. In preparation.
- Bąkowska-Żywicka, K.; Kasprzyk, M.; Twardowski, T. tRNA-derived short RNAs bind to Saccharomyces cerevisiae ribosomes in a stress-dependent manner and inhibit protein synthesis in vitro. FEMS Yeast Res. 2016, 16, fow077. [Google Scholar] [CrossRef]
- Potapov, V.; Fu, X.; Dai, N.; Corrêa, I.R., Jr.; Tanner, N.A.; Ong, J.L. Base modifications affecting RNA polymerase and reverse transcriptase fidelity. Nucleic Acids Res. 2018, 46, 5753–5763. [Google Scholar] [CrossRef]
- Su, Z.; Wilson, B.; Kumar, P.; Dutta, A. Noncanonical Roles of tRNAs: tRNA Fragments and Beyond. Annu. Rev. Genet. 2020, 54, 47–69. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Tesina, P.; Nakajima, S.; Mizuno, M.; Endo, A.; Buschauer, R.; Cheng, J.; Shounai, O.; Ikeuchi, K.; Saeki, Y.; et al. RQT complex dissociates ribosomes collided on endogenous RQC substrate SDD1. Nat. Struct. Mol. Biol. 2020, 27, 323–332. [Google Scholar] [CrossRef]
- Bao, C.; Ermolenko, D.N. Ribosome as a Translocase and Helicase. Biochemistry 2021, 86, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- West, M.; Zurek, N.; Hoenger, A.; Voeltz, G.K. A 3D analysis of yeast ER structure reveals how ER domains are organized by membrane curvature. J Cell Biol. 2011, 193, 333–346. [Google Scholar] [CrossRef]
- Favre, A.; Saintomé, C.; Fourrey, J.L.; Clivio, P.; Laugâa, P. Thionucleobases as intrinsic photoaffinity probes of nucleic acid structure and nucleic acid-protein interactions. J. Photochem. Photobiol. B Biol. 1998, 42, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Byrd, A.K.; Zybailov, B.L.; Marecki, J.C.; Guderyon, M.J.; Edwards, A.D.; Chib, S.; West, K.L.; Waldrip, Z.J.; Mackintosh, S.G.; et al. DEAD-box RNA helicases Dbp2, Ded1 and Mss116 bind to G-quadruplex nucleic acids and destabilize G-quadruplex RNA. Chem. Commun. 2019, 55, 4467–4470. [Google Scholar] [CrossRef] [PubMed]
- Herdy, B.; Mayer, C.; Varshney, D.; Marsico, G.; Murat, P.; Taylor, C.; D′Santos, C.; Tannahill, D.; Balasubramanian, S. Analysis of NRAS RNA G-quadruplex binding proteins reveals DDX3X as a novel interactor of cellular G-quadruplex containing transcripts. Nucleic Acids Res. 2018, 46, 11592–11604. [Google Scholar] [CrossRef] [PubMed]
- Čutová, M.; Manta, J.; Porubiaková, O.; Kaura, P.; Šťastný, J.; Jagelská, E.B.; Goswami, P.; Bartas, M.; Brázda, V. Divergent distributions of inverted repeats and G-quadruplex forming sequences in Saccharomyces cerevisiae. Genomics 2020, 112, 1897–1901. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.D.; Zhou, F.; Ingolia, N.T.; Hinnebusch, A.G. Genome-wide analysis of translational efficiency reveals distinct but overlapping functions of yeast DEAD-box RNA helicases Ded1 and eIF4A. Genome Res. 2015, 25, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- Collart, M.A.; Weiss, B. Ribosome pausing, a dangerous necessity for co-translational events. Nucleic Acids Res. 2020, 48, 1043–1055. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | ORF | 5′ UTR | 3′ UTR | GO Term a |
|---|---|---|---|---|
| TAR1 | 374 | 110 | 91 | Cellular respiration (GO:0045333) |
| DDR2 | 185 | 34 | 244 | Response to oxidative stress (GO:0006979) |
| HKR1 | 5408 | 31 | 142 | Response to osmotic stress (GO:0006970) |
| HSP26 | 644 | 60 | 165 | Protein folding (GO:0006457) |
| SOL4 | 767 | 35 | 96 | Generation of precursor metabolites and energy (GO:0006091) |
| TKL2 | 2045 | – 2 | – 2 | Generation of precursor metabolites and energy (GO:0006091) |
| GLK1 | 1502 | 31 | 180 | Generation of precursor metabolites and energy (GO:0006091) |
| GSY1 | 2126 | 70 | 112 | Generation of precursor metabolites and energy (GO:0006091) |
| TEF1 | 1376 | 33 | 123 | Translational elongation (GO:0006414) |
| YSC84 | 1574 | 47 | 109 | Cytoskeleton organization (GO:0007010) |
| SPI1 | 446 | 21 | 165 | GPI-anchored cell wall protein |
| GND2 | 1478 | – 2 | 114 | Generation of precursor metabolites and energy (GO:0006091) |
| AFG1 | 1529 | 88 | 61 | Response to oxidative stress (GO:0006979) |
| HST3 | 1343 | 43 | 149 | Chromatin organization (GO:0006325) |
| STF1 | 260 | 53 | 164 | Regulator of mitochondrial F1F0-ATP synthase |
| ALD4 | 1559 | 39 | 226 | Generation of precursor metabolites and energy (GO:0006091) |
| APE3 | 1613 | 33 | 50 | Vacuolar aminopeptidase Y |
| SIL1 | 1265 | 63 | 23 | Protein targeting (GO:0006605) |
| HBT1 | 3140 | – 2 | – 2 | Cell morphogenesis (GO:0000902) |
| GAD1 | 1757 | 39 | 105 | Amino acid metabolic process (GO:0006520) |
| Gene | ORF | 5′ UTR | 3′ UTR | GO Term a |
|---|---|---|---|---|
| CCT7 | 1652 | 166 | 130 | Protein folding (GO:0006457) |
| BFR1 | 1412 | – 2 | 200 | Regulation of organelle organization (GO:0033043) |
| RPL31A | 762 | 21 | 70 | Cytoplasmic translation (GO:0002181) |
| VMA2 | 1553 | 46 | 148 | Transmembrane transport (GO:0055085) |
| GDH1 | 1364 | 57 | 81 | Amino acid metabolic process (GO:0006520) |
| XRN1 | 4586 | 333 | 124 | RNA catabolic process (GO:0006401) |
| RRP14 | 1304 | 74 | 91 | Ribosome assembly (GO:0042255) |
| PBP1 | 2168 | 215 | 101 | Regulation of translation (GO:0006417) |
| RPS14A | 720 | 22 | 78 | rRNA processing (GO:0006364) |
| TSR1 | 2366 | 42 | 98 | rRNA processing (GO:0006364) |
| USO1 | 5372 | 33 | 136 | Vesicle organization (GO:0016050) |
| NUM1 | 8246 | – 2 | 50 | Cytoskeleton organization (GO:0007010) |
| ENP2 | 2123 | 48 | 35 | rRNA processing (GO:0006364) |
| MPP10 | 1781 | 17 | 361 | rRNA processing (GO:0006364) |
| CAB4 | 917 | 19 | 53 | Small molecule metabolic process (GO:0055086) |
| SUB2 | 1340 | 156 | 111 | RNA splicing (GO:0008380) |
| SEC62 | 824 | 12 | 44 | Protein targeting (GO:0006605) |
| TUB2 | 1373 | 66 | 226 | Cytoskeleton organization (GO:0007010) |
| SEC61 | 1442 | 206 | 144 | Protein targeting (GO:0006605) |
| MAL31 | 1844 | 155 | 69 | Maltose permease |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeter-Alat, H.; Belgareh-Touzé, N.; Huvelle, E.; Banroques, J.; Tanner, N.K. The DEAD-Box RNA Helicase Ded1 Is Associated with Translating Ribosomes. Genes 2023, 14, 1566. https://doi.org/10.3390/genes14081566
Yeter-Alat H, Belgareh-Touzé N, Huvelle E, Banroques J, Tanner NK. The DEAD-Box RNA Helicase Ded1 Is Associated with Translating Ribosomes. Genes. 2023; 14(8):1566. https://doi.org/10.3390/genes14081566
Chicago/Turabian StyleYeter-Alat, Hilal, Naïma Belgareh-Touzé, Emmeline Huvelle, Josette Banroques, and N. Kyle Tanner. 2023. "The DEAD-Box RNA Helicase Ded1 Is Associated with Translating Ribosomes" Genes 14, no. 8: 1566. https://doi.org/10.3390/genes14081566
APA StyleYeter-Alat, H., Belgareh-Touzé, N., Huvelle, E., Banroques, J., & Tanner, N. K. (2023). The DEAD-Box RNA Helicase Ded1 Is Associated with Translating Ribosomes. Genes, 14(8), 1566. https://doi.org/10.3390/genes14081566