The Novel Variant NP_00454563.2 (p.Glu259Glyfs*77) in Gene PKP2 Associated with Arrhythmogenic Cardiomyopathy in 8 Families from Malaga, Spain

 , , and
, , and
Abstract
1. Introduction
2. Methods
2.1. Study Design and Population
2.2. Genetic Analysis
2.3. Clinical Evaluation and Follow-Up
2.4. Statistical Analysis
3. Results
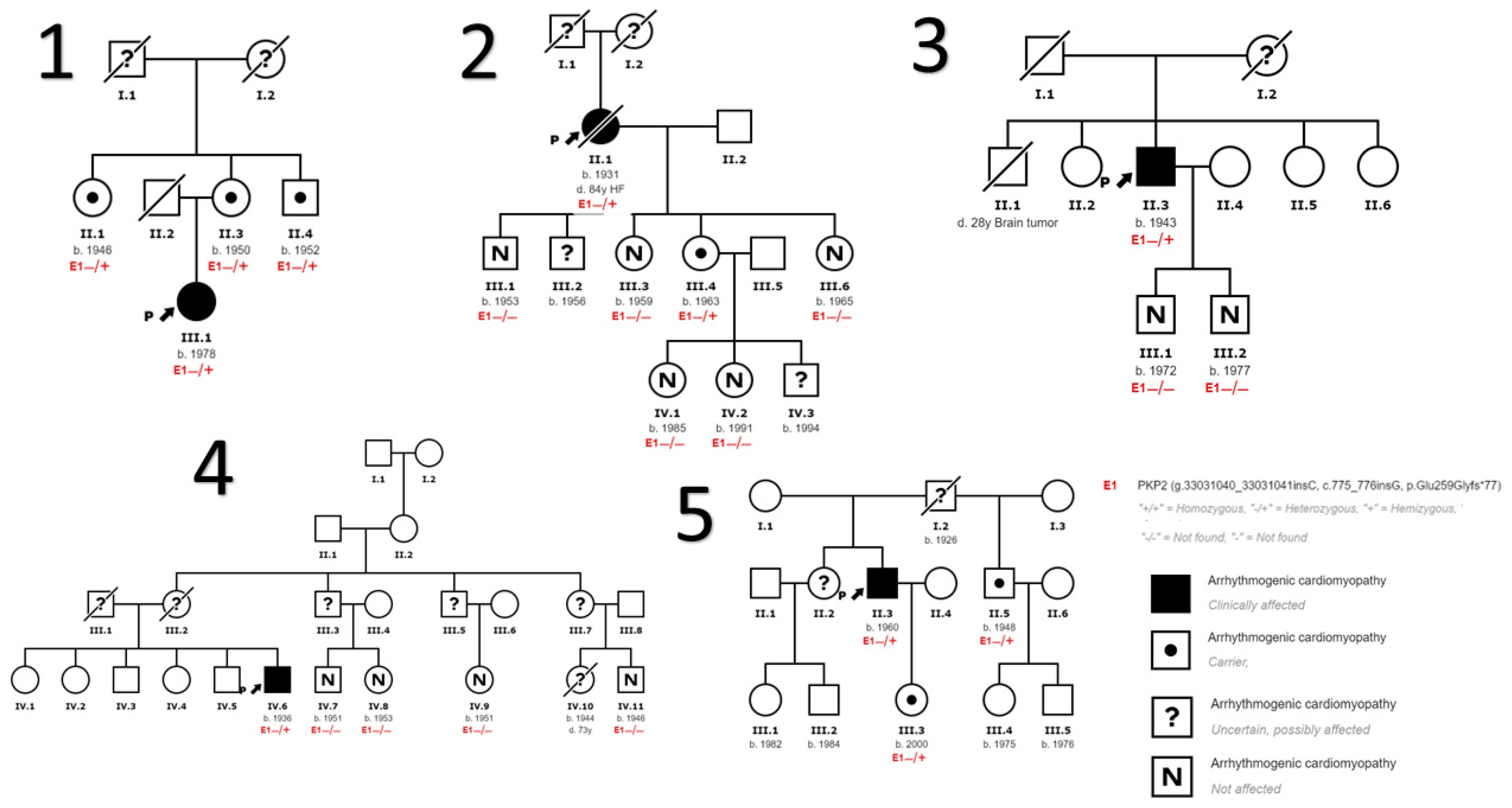
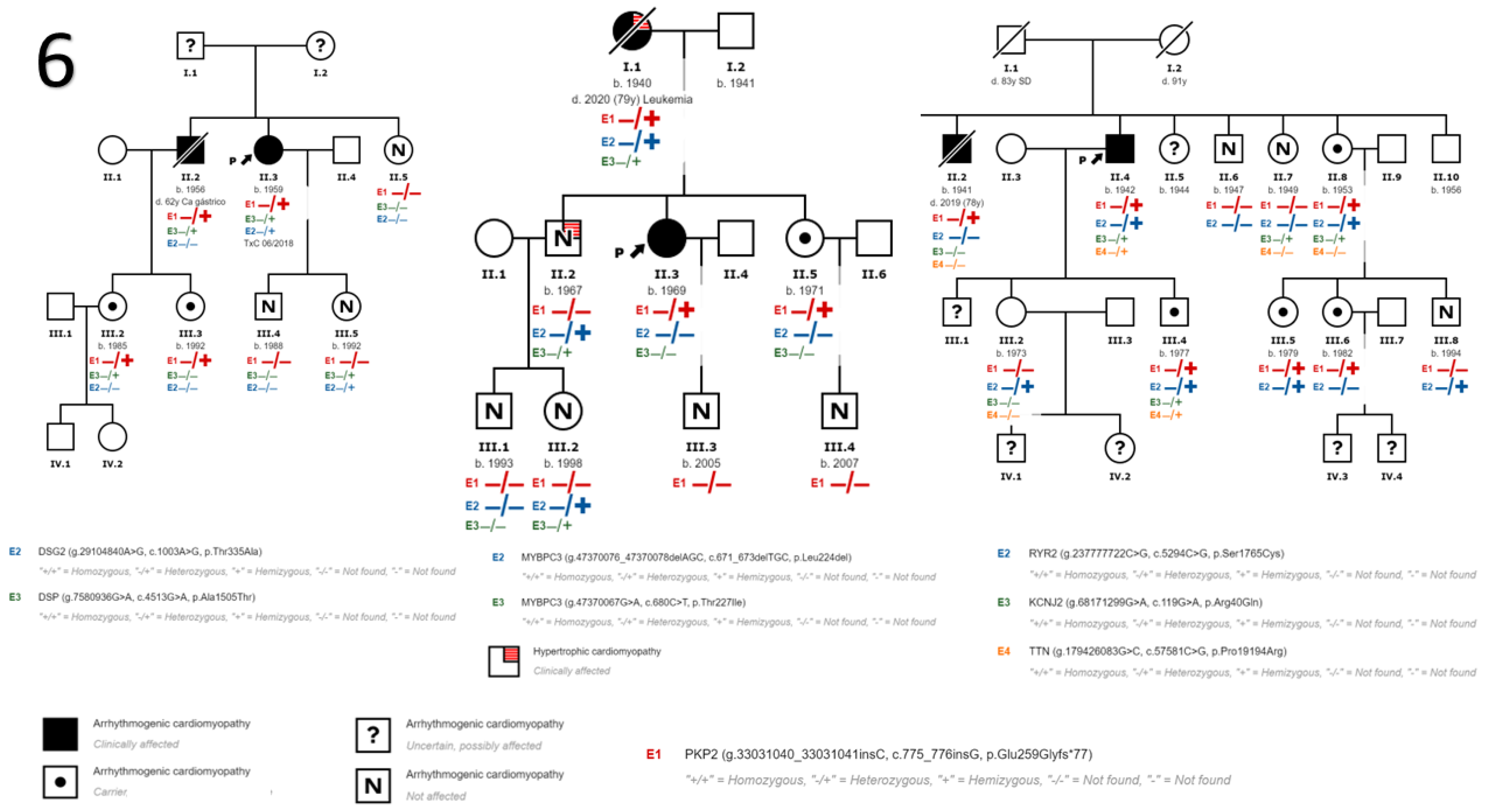
3.1. Baseline Characteristics of the Sample
3.2. Follow-Up
4. Discussion
Limitations
- -
- ACM is a heart disease defined by the progressive replacement of the right ventricle myocardium with fibroadipose tissue, and may cause arrhythmias, sudden death, and heart failure. Currently, 40–60% of patients have at least one genetic mutation related with the disease.
- -
- This study presents a PKP2 variant not previously described or present in the control population, and establishes a probable founder effect in our region.
- -
- This variant has incomplete penetrance and a highly variable phenotypic expressivity, and is strongly influenced by age, sex, and the presence of other genetic or environmental factors such as sport, characteristics that the majority of desmosomal gene mutations share.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACM | arrhythmogenic cardiomyopathy |
| ARVD | arrhythmogenic right ventricular dysplasia |
| CMRI | cardiac magnetic resonance imaging |
| DCM | dilated cardiomyopathy |
| DSC2 | desmocollin-2 |
| DSG2 | desmoglein-2 |
| DSP | desmoplakin |
| HCM | hypertrophic cardiomyopathy |
| HF | heart failure |
| ICD | implantable cardioverter–defibrillator |
| JUP | junction plakoglobin |
| KCNJ2 | inward-rectifier potassium |
| LV | left ventricle |
| LVEF | left ventricular ejection fraction |
| MYBPC3 | myosin binding protein C3 |
| NGS | next-generation sequencing |
| NSVT | non-sustained ventricular tachycardia |
| PKP2 | plakophilin-2 |
| RV | right ventricle |
| RYR2 | ryanodine receptor 2 |
| SD | sudden death |
| SVT | sustained ventricular tachycardia |
| TTN | titin |
| VPB | ventricular premature beats |
References
- Basso, C.; Corrado, D.; Thiene, G. Cardiovascular causes of sudden death in young individuals including athletes. Cardiol. Rev. 1999, 7, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Marcus, F.I.; Fontaine, G.H.; Guiraudon, G.H.; Frank, R.; Laurenceau, J.L.; Malergue, C.; Grosgogeat, Y. Right ventricular dysplasia: A report of 24 adult cases. Circulation 1982, 65, 384–398. [Google Scholar] [CrossRef] [PubMed]
- López-Moreno, E.; Jiménez-Jáimez, J.; Macías-Ruiz, R.; Sánchez-Millán, P.J.; Álvarez-López, M.; Tercedor-Sánchez, L. Clinical Profile of Arrhythmogenic Right Ventricular Cardiomyopathy With Left Ventricular Involvement. Rev. Española Cardiol. 2016, 69, 872–874. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Syrris, P.; Prasad, S.K.; Hughes, S.E.; Merrifield, R.; Ward, D.; Pennell, D.J.; McKenna, W.J. Left-Dominant Arrhythmogenic Cardiomyopathy. An Under-Recognized Clinical Entity. J. Am. Coll. Cardiol. 2008, 52, 2175–2187. [Google Scholar] [CrossRef] [PubMed]
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef]
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic cardiomyopathy. Circ. Res. 2017, 121, 785–802. [Google Scholar] [CrossRef]
- Quarta, G.; Elliott, P.M. Diagnostic criteria for arrhythmogenic right ventricular cardiomyopathy. Rev. Esp. Cardiol. 2012, 65, 599–605. [Google Scholar] [CrossRef]
- Ackerman, M.J.; Priori, S.G.; Willems, S.; Berul, C.; Brugada, R.; Calkins, H.; Camm, A.J.; Ellinor, P.T.; Gollob, M.H.; Hamilton, R.M.; et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Europace 2011, 13, 1077–1109. [Google Scholar] [CrossRef]
- Protonotarios, N.; Tsatsopoulou, A. Naxos disease and Carvajal syndrome: Cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc. Pathol. 2004, 13, 185–194. [Google Scholar] [CrossRef]
- James, C.A.; Syrris, P.; Van Tintelen, J.P.; Calkins, H. The role of genetics in cardiovascular disease: Arrhythmogenic cardiomyopathy. Eur. Heart J. 2020, 41, 1393–1400. [Google Scholar] [CrossRef]
- James, C.A.; Jongbloed, J.D.; Hershberger, R.E.; Morales, A.; Judge, D.P.; Syrris, P.; Pilichou, K.; Domingo, A.M.; Murray, B.; Cadrin-Tourigny, J.; et al. International Evidence Based Reappraisal of Genes Associated with Arrhythmogenic Right Ventricular Cardiomyopathy Using the Clinical Genome Resource Framework. Circ. Genom. Precis Med. 2021, 14, e003273. [Google Scholar] [CrossRef]
- Corrado, D.; Van Tintelen, P.J.; McKenna, W.J.; Hauer, R.N.W.; Anastastakis, A.; Asimaki, A.; Basso, C.; Bauce, B.; Brunckhorst, C.; Bucciarelli-Ducci, C.; et al. Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria and differential diagnosis. Eur. Heart. J. 2020, 41, 1414–1429. [Google Scholar] [CrossRef]
- Kapplinger, J.D.; Landstrom, A.P.; Salisbury, B.A.; Callis, T.E.; Pollevick, G.D.; Tester, D.J.; Cox, M.G.; Bhuiyan, Z.; Bikker, H.; Wiesfeld, A.C.; et al. Distinguishing arrhythmogenic right ventricular cardiomyopathy/dysplasia- associated mutations from background genetic noise. J. Am. Coll Cardiol. 2011, 57, 2317–2327. [Google Scholar] [CrossRef]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur. Heart J. 2010, 31, 806–814. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.; Ackerman, M.J.; Calkins, H.; Darrieux, F.; Daubert, J.P.; De Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019, 16, e301–e372. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Jacob, K.A.; Noorman, M.; Cox, M.G.P.J.; Groeneweg, J.A.; Hauer, R.N.W.; van der Heyden, M.A.G. Geographical distribution of plakophilin-2 mutation prevalence in patients with arrhythmogenic cardiomyopathy. Neth. Heart J. 2012, 20, 234–239. [Google Scholar] [CrossRef]
- Groeneweg, J.A.; Bhonsale, A.; James, C.A.; Te Riele, A.S.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfeld, A.C.; Sawant, A.C.; Kassamali, B.; et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ. Cardiovasc. Genet. 2015, 8, 437–446. [Google Scholar] [CrossRef]
- Cox, M.G.; van der Zwaag, P.A.; van der Werf, C.; van der Smagt, J.J.; Noorman, M.; Bhuiyan, Z.A.; Wiesfeld, A.C.; Volders, P.G.; van Langen, I.M.; Atsma, D.E.; et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: Pathogenic desmosome mutations in index-patients predict outcome of family screening: Dutch arrhythmogenic right ventricular dysplasia/cardiomyopathy genotype-phenotype follow-up study. Circulation 2011, 123, 2690–2700. [Google Scholar] [CrossRef]
- Van Lint, F.H.; Murray, B.; Tichnell, C.; Zwart, R.; Amat, N.; Deprez, R.H.L.; Dittmann, S.; Stallmeyer, B.; Calkins, H.; van der Smagt, J.J.; et al. Arrhythmogenic Right Ventricular Cardiomyopathy-Associated Desmosomal Variants Are Rarely De Novo. Circ. Genom. Precis. Med. 2019, 12, e002467. [Google Scholar] [CrossRef] [PubMed]
- Alcalde, M.; Campuzano, O.; Sarquella-Brugada, G.; Arbelo, E.; Allegue, C.; Partemi, S.; Iglesias, A.; Oliva, A.; Brugada, J.; Brugada, R. Clinical interpretation of genetic variants in arrhythmogenic right ventricular cardiomyopathy. Clin. Res. Cardiol. 2015, 104, 288–303. [Google Scholar] [CrossRef] [PubMed]
- Ruiz Salas, A.; Hernández, J.P.; Palomo, C.M.; Cordero, A.B.; Bueno, F.C.; Pinilla, J.M.G.; Guijarro, A.; Morcillo-Hidalgo, L.; Navarro, M.J.; Doblas, J.J.G.; et al. Usefulness of Genetic Study by Next-generation Sequencing in High-risk Arrhythmogenic Cardiomyopathy. Rev. Española Cardiol. 2018, 71, 1018–1026. [Google Scholar] [CrossRef]
- Christensen, A.H.; Benn, M.; Bundgaard, H.; Tybjærg-Hansen, A.; Haunso, S.; Svendsen, J.H. Wide spectrum of desmosomal mutations in Danish patients with arrhythmogenic right ventricular cardiomyopathy. J. Med. Genet. 2010, 47, 736–744. [Google Scholar] [CrossRef]
- Den Haan, A.D.; Tan, B.Y.; Zikusoka, M.N.; Lladó, L.I.; Jain, R.; Daly, A.; Tichnell, C.; James, C.; Amat-Alarcon, N.; Abraham, T.; et al. Comprehensive desmosome mutation analysis in North Americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ. Cardiovasc. Genet. 2009, 2, 428–435. [Google Scholar] [CrossRef]
- Fressart, V.; Duthoit, G.; Donal, E.; Probst, V.; Deharo, J.-C.; Chevalier, P.; Klug, D.; Dubourg, O.; Delacretaz, E.; Cosnay, P.; et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: Spectrum of mutations and clinical impact in practice. Europace 2010, 12, 861–868. [Google Scholar] [CrossRef]
- Van Tintelen, J.P.; Entius, M.M.; Bhuiyan, Z.A.; Jongbloed, R.; Wiesfeld, A.C.; Wilde, A.A.; Boven, L.G.; Mannens, M.M.; van Langen, I.M.; Hofstra, R.M.; et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation 2006, 113, 1650–1658. [Google Scholar] [CrossRef]
- Dalal, D.; James, C.; Devanagondi, R.; Tichnell, C.; Tucker, A.; Prakasa, K.; Spevak, P.J.; Bluemke, D.A.; Abraham, T.; Russell, S.D.; et al. Penetrance of Mutations in Plakophilin-2 Among Families With Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy. J. Am. Coll. Cardiol. 2006, 48, 1416–1424. [Google Scholar] [CrossRef]
- Dalal, D.; Molin, L.H.; Piccini, J.; Tichnell, C.; James, C.; Bomma, C.; Prakasa, K.; Towbin, J.A.; Marcus, F.I.; Spevak, P.J.; et al. Clinical features of arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in plakophilin-2. Circulation 2006, 113, 1641–1649. [Google Scholar] [CrossRef]
- Bhonsale, A.; Groeneweg, J.A.; James, C.A.; Dooijes, D.; Tichnell, C.; Jongbloed, J.D.H.; Murray, B.; Te Riele, A.S.J.M.; Van Den Berg, M.P.; Bikker, H.; et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur. Heart J. 2015, 36, 847–855. [Google Scholar] [CrossRef]
- Bao, J.; Wang, J.; Yao, Y.; Wang, Y.; Fan, X.; Sun, K.; He, D.S.; Marcus, F.I.; Zhang, S.; Hui, R.; et al. Correlation of ventricular arrhythmias with genotype in arrhythmogenic right ventricular cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 552–556. [Google Scholar] [CrossRef]
- Chivulescu, M.; Lie, H.; Popescu, A.B.; Skulstad, H.; Edvardsen, T.; Jurcut, O.R.; Haugaa, K.H. High penetrance and similar disease progression in probands and in family members with arrhythmogenic cardiomyopathy. Eur. Heart J. 2020, 41, 1401–1410. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| A. | Carriers | Non-Carriers | p | Total |
|---|---|---|---|---|
| Sex | 0.38 | |||
| Women | 15 (31.9%) | 11 (23.4%) | 26 (55.3%) | |
| Men | 9 (19.1%) | 12 (25.5%) | 21 (44.7%) | |
| Age (years) | 54 ± 16.7 | 43.6 ± 19.3 | 0.06 | 48.9 ± 18.6 |
| Follow-up time (months) | 48 [27–59] | 27 [23–59] | 0.13 | 39 [24–59] |
| LVEF (%) | 63 [45–65] | 64 [62–65] | 0.43 | 64 [60–65] |
| B. | Men | Women | p | Total |
| Diagnostic ACM/DCM | 6 (54.5%) | 5 (45.5%) | 0.50 | 11 (100%) |
| Probands | 4 (50%) | 4 (50%) | 8 (72.7%) | |
| Family members | 2 (66.7%) | 1 (33.3%) | 3 (27.2%) | |
| Age at diagnosis (years) | 63.2 ± 11.2 | 48.4 ± 17.3 | 0.03 | 54 ± 16.7 |
| Form of presentation | 0.26 | |||
| Family screening | 2 (18.2%) | 0 (0%) | 2 (18.2%) | |
| Dyspnoea | 1 (9.1%) | 2 (18.2%) | 3 (27.2%) | |
| Palpitations | 0 (0%) | 1 (9.1%) | 1 (9.1%) | |
| Arrhythmic events | 3 (27.2%) | 1 (9.1%) | 4 (36.3%) | |
| Sudden death | 0 (0%) | 1 (9.1%) | 1 (9.1%) | |
| Involvement | 0.32 | |||
| Right | 3 (27.2%) | 1 (9.1%) | 4 (36.3%) | |
| Left | 2 (18.2%) | 1 (9.1%) | 3 (27.2%) | |
| Biventricular | 1 (9.1%) | 3 (27.1%) | 4 (36.3%) | |
| ECG | 0.47 | |||
| RBBB | 3 (27.2%) | 3 (27.2%) | 6 (54.4%) | |
| T negative wave | 1 (9.1%) | 2 (18.2%) | 3 (27.2%) | |
| Epsilon wave | 1 (9.1%) | 0 (0%) | 1 (9.1%) | |
| LVEF (%) | 63 [40–65] | 64 [60–65] | 0.68 | 63 [45–65] |
| Follow-up time (months) | 59 [33–75] | 42 [24–58] | 0.07 | 48 [27–59] |
| ICD implant | 0.25 | |||
| Primary prevention | 1 (9.1%) | 3 (27.2%) | 4 (36.3%) | |
| Secondary prevention | 2 (18.2%) | 2 (18.2%) | 4 (36.3%) | |
| Events in the follow-up | 0.45 | |||
| SVT | 2 (18.2%) | 2 (18.2%) | 4 (36.3%) | |
| NSVT | 0 (0%) | 1 (9.1%) | 1 (9.1%) | |
| VT ablation | 1 (9.1%) | 1 (9.1%) | 2 (27.2%) | |
| ICD therapies | 1 (9.1%) | 1 (9.1%) | 2 (27.2%) | |
| HF | 3 (27.2%) | 3 (27.2%) | 6 (54.5%) | |
| Heart transplantation | 0 (0%) | 1 (9.1%) | 1 (9.1%) |
| Family/ Patient | Sex | Age 1st Visit (Years) | Index Case | Other Genetic Variants | Presentation | Involvement | LVEF (%) | ICD | Arrhythmias under Follow-Up | HF | Death |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1/III.1 | Woman | 34 | Yes | No | Palpitations (VPB) | Right | 65 | Primary Prevention | SVT with focus ablation | No | No |
| 2/II.1 | Woman | 76 | Yes | No | VT | Biventricular | 38 | Secondary Prevention | SVT | Yes | Yes (HF) |
| 3/II.3 | Man | 69 | Yes | No | VT | Right | 65 | Secondary Prevention | SVT | No | No |
| 4/IV.6 | Man | 73 | Yes | No | VT | Right | 65 | No | No | No | No |
| 5/II.3 | Man | 54 | Yes | No | VT | Right | 65 | Secondary Prevention | SVT with focus ablation | No | No |
| 6/II.3 | Woman | 56 | Yes | DSP p.Ala1505Thr (+?) DSG2 p.Thr335Al (+++) | Dyspnea | Left | 30 | Primary Prevention | NSVT | Yes (CT) | No |
| 6/II.2 | Man | 63 | No | DSP p.Ala1505Thr (+?) | Family Study | Left | 40 | No | No | Yes | Yes (gastric cancer) |
| 7/II.3 | Woman | 48 | Yes | No | SD | Biventricular | 65 | Secondary Prevention | No | No | No |
| 7/I.1 | Woman | 65 | No | MYBPC3 p.Leu224del (++) * MYBPC3 p.Thr227Ile (++) * | Dyspnea | Biventricular | 38 | Primary Prevention | No | Yes | Yes (leukaemia) |
| 8/II.4 | Man | 63 | Yes | RYR2 p.Ser1765Cys (+?) KCNJ2 p.Arg40Gln (-?) TTN p.Pro19194Arg (-?) | Dyspnea | Left | 40 | No | No | Yes | No |
| 8/II.2 | Man | 74 | No | RYR2 p.Ser1765Cys (+?) KCNJ2 p.Arg40Gln (-?) TTN p.Pro19194Arg (-?) | Family Study | Biventricular | 30 | Primary Prevention | No | Yes | Yes (older age) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robles-Mezcua, A.; Ruíz-Salas, A.; Medina-Palomo, C.; Robles-Mezcua, M.; Díaz-Expósito, A.; Ortega-Jiménez, M.V.; Gimeno-Blanes, J.R.; Jiménez-Navarro, M.F.; García-Pinilla, J.M. The Novel Variant NP_00454563.2 (p.Glu259Glyfs*77) in Gene PKP2 Associated with Arrhythmogenic Cardiomyopathy in 8 Families from Malaga, Spain. Genes 2023, 14, 1468. https://doi.org/10.3390/genes14071468
Robles-Mezcua A, Ruíz-Salas A, Medina-Palomo C, Robles-Mezcua M, Díaz-Expósito A, Ortega-Jiménez MV, Gimeno-Blanes JR, Jiménez-Navarro MF, García-Pinilla JM. The Novel Variant NP_00454563.2 (p.Glu259Glyfs*77) in Gene PKP2 Associated with Arrhythmogenic Cardiomyopathy in 8 Families from Malaga, Spain. Genes. 2023; 14(7):1468. https://doi.org/10.3390/genes14071468
Chicago/Turabian StyleRobles-Mezcua, Ainhoa, Amalio Ruíz-Salas, Carmen Medina-Palomo, María Robles-Mezcua, Arancha Díaz-Expósito, María Victoria Ortega-Jiménez, Juan Ramón Gimeno-Blanes, Manuel F. Jiménez-Navarro, and José Manuel García-Pinilla. 2023. "The Novel Variant NP_00454563.2 (p.Glu259Glyfs*77) in Gene PKP2 Associated with Arrhythmogenic Cardiomyopathy in 8 Families from Malaga, Spain" Genes 14, no. 7: 1468. https://doi.org/10.3390/genes14071468
APA StyleRobles-Mezcua, A., Ruíz-Salas, A., Medina-Palomo, C., Robles-Mezcua, M., Díaz-Expósito, A., Ortega-Jiménez, M. V., Gimeno-Blanes, J. R., Jiménez-Navarro, M. F., & García-Pinilla, J. M. (2023). The Novel Variant NP_00454563.2 (p.Glu259Glyfs*77) in Gene PKP2 Associated with Arrhythmogenic Cardiomyopathy in 8 Families from Malaga, Spain. Genes, 14(7), 1468. https://doi.org/10.3390/genes14071468