
Characterization and Potential Function Analysis of the SRS Gene Family in Brassica napus

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Characterization and Physicochemical Characterization of BnSRS Family Members
2.2. The Phylogenetic Analysis of BnSRS
2.3. The Gene Structure, Conserved Motifs, and Cis-Acting Regulatory Elements Analysis
2.4. Analysis of the Expression Pattern of BnSRS in Different Tissues and Different Environments
2.5. Prediction of Protein-Protein Interaction Network Analysis
2.6. Association Mapping Analysis of BnSRS Genes with Important Agronomic Traits
3. Results and Discussion
3.1. Identification of BnSRS Genes and Analysis of Physicochemical Properties of Its Family Members
3.2. Phylogenetic Analysis of BnSRS
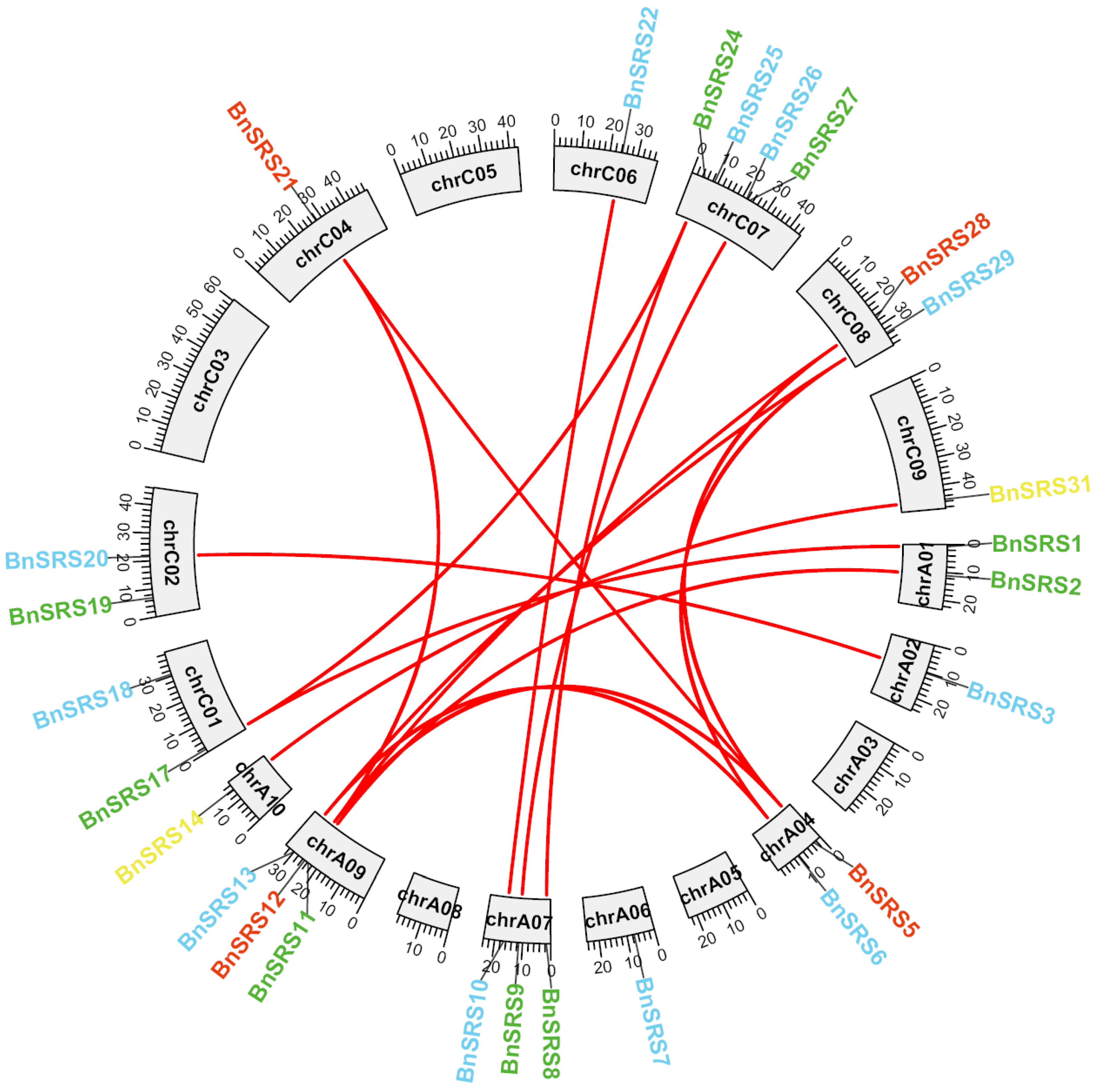
3.3. Analysis of Chromosomal Localization and Duplication Events of BnSRS Gene Family Members
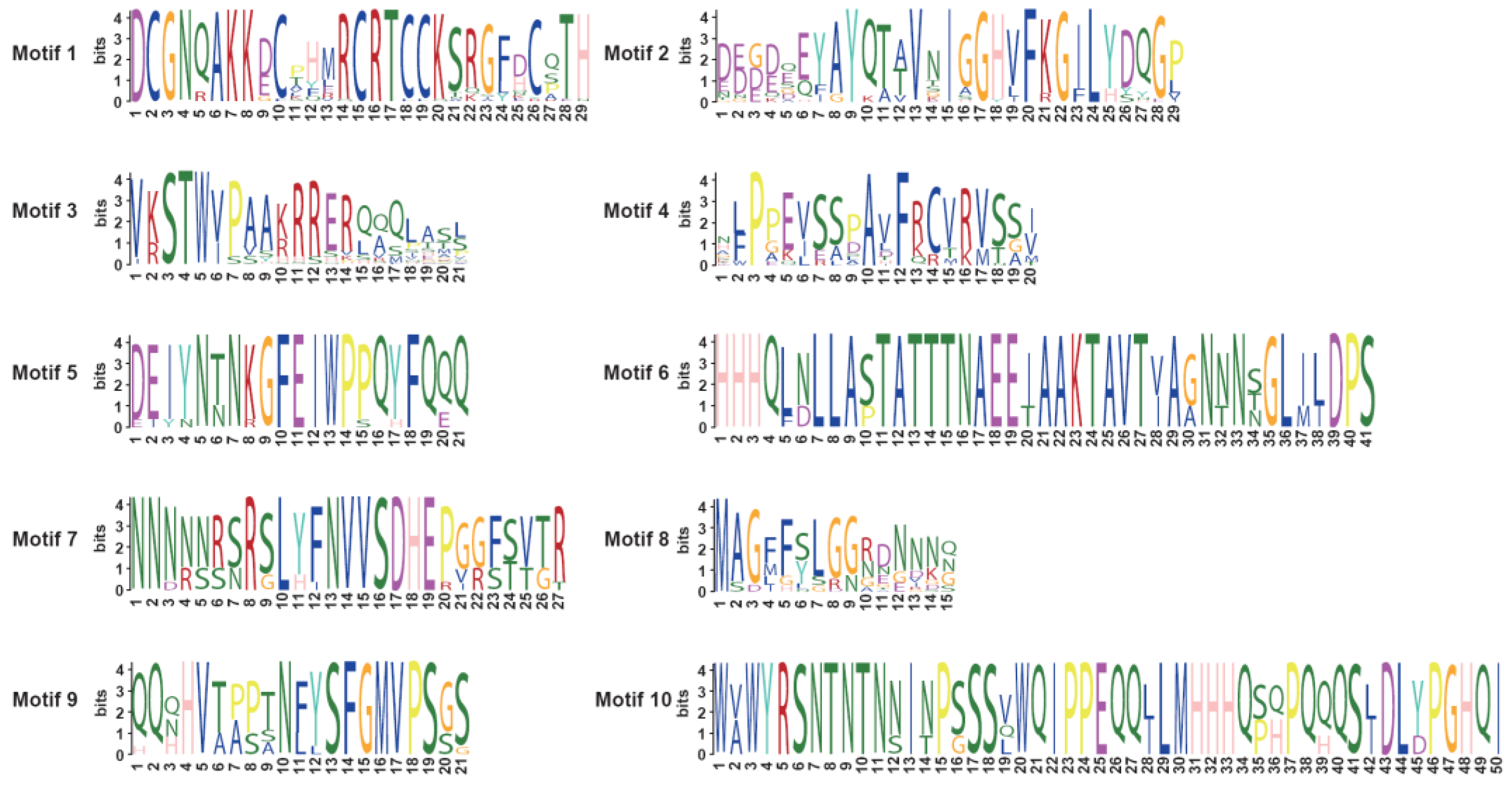
3.4. Analysis of BnSRS Gene Structure and Conserved Motifs
3.5. Analysis of Cis-Acting Elements of the BnSRS Genes
3.6. Expression Pattern Analysis of BnSRS Gene Family Members
3.7. Prediction of BnSRS Proteins Interactions Analysis
3.8. Genetic Effects of BnSRS Genes on B. napus Agronomic Traits
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Yadav, S.; Singh, A.; Mahima, M.; Singh, A.; Gautam, V.; Sarkar, A.K. Auxin signaling modulates LATERAL ROOT PRIMORDIUM1 (LRP1) expression during lateral root development in Arabidopsis. Plant J. 2020, 101, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.L.; Fedoroff, N.V. LRP1, a gene expressed in lateral and adventitious root primordia of arabidopsis. Plant Cell 1995, 7, 735–745. [Google Scholar]
- De Rybel, B.; Audenaert, D.; Xuan, W.; Overvoorde, P.; Strader, L.C.; Kepinski, S.; Hoye, R.; Brisbois, R.; Parizot, B.; Vanneste, S.; et al. A role for the root cap in root branching revealed by the non-auxin probe naxillin. Nat. Chem. Biol. 2012, 8, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Eklund, D.M.; Cierlik, I.; Ståldal, V.; Claes, A.R.; Vestman, D.; Chandler, J.; Sundberg, E. Expression of Arabidopsis SHORT INTERNODES/STYLISH family genes in auxin biosynthesis zones of aerial organs is dependent on a GCC box-like regulatory element. Plant Physiol. 2011, 157, 2069–2080. [Google Scholar] [CrossRef] [PubMed]
- Gomariz-Fernández, A.; Sánchez-Gerschon, V.; Fourquin, C.; Ferrándiz, C. The Role of SHI/STY/SRS Genes in Organ Growth and Carpel Development Is Conserved in the Distant Eudicot Species Arabidopsis thaliana and Nicotiana benthamiana. Front. Plant Sci. 2017, 8, 814. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, P.; Yu, D. Genome-Wide Identification and Characterization of the SHI-Related Sequence Gene Family in Rice. Evol. Bioinform. Online 2020, 16, 1176934320941495. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Yu, L.; Zhang, S.; Gu, Q.; Wang, M. Genome-wide characterization of the SHORT INTER-NODES/STYLISH and Shi-Related Sequence family in Gossypium hirsutum and functional identification of GhSRS21 under salt stress. Front. Plant Sci. 2022, 13, 1078083. [Google Scholar] [CrossRef]
- Zhao, S.P.; Song, X.Y.; Guo, L.L.; Zhang, X.Z.; Zheng, W.J. Genome-Wide Analysis of the Shi-Related Sequence Family and Functional Identification of GmSRS18 Involving in Drought and Salt Stresses in Soybean. Int. J. Mol. Sci. 2020, 21, 1810. [Google Scholar] [CrossRef]
- Kuusk, S.; Sohlberg, J.J.; Long, J.A.; Fridborg, I.; Sundberg, E. STY1 and STY2 promote the formation of apical tissues during Arabidopsis gynoecium development. Development 2002, 129, 4707–4717. [Google Scholar] [CrossRef]
- Fridborg, I.; Kuusk, S.; Robertson, M.; Sundberg, E. The Arabidopsis protein SHI represses gibberellin responses in Arabidopsis and barley. Plant Physiol. 2001, 127, 937–948. [Google Scholar] [CrossRef]
- Kuusk, S.; Sohlberg, J.J.; Magnus Eklund, D.; Sundberg, E. Functionally redundant SHI family genes regulate Arabidopsis gynoecium development in a dose-dependent manner. Plant J. 2006, 47, 99–111. [Google Scholar] [CrossRef]
- Eklund, D.M.; Ståldal, V.; Valsecchi, I.; Cierlik, I.; Eriksson, C.; Hiratsu, K.; Ohme-Takagi, M.; Sundström, J.F.; Thelander, M.; Ezcurra, I.; et al. The Arabidopsis thaliana STYLISH1 protein acts as a transcriptional activator regulating auxin biosynthesis. Plant Cell 2010, 22, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Kulkarni, V.; Liu, N.; Del Carpio, D.P.; Bucher, J.; Bonnema, G. BrFLC2 (FLOWERING LOCUS C) as a candidate gene for a vernalization response QTL in Brassica rapa. J. Exp. Bot. 2010, 61, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.-G.; Zhao, Z.-Q.; Wang, J.-S.; Yu, H.-F.; Shen, Y.-S.; Zeng, X.-Y.; Gu, H.-H. Genome wide analysis of MADS-box gene family in Brassica oleracea reveals conservation and variation in flower development. BMC Plant Biol. 2019, 19, 106. [Google Scholar] [CrossRef] [PubMed]
- Treccarichi, S.; Di Gaetano, C.; Di Stefano, F.; Gasparini, M.; Branca, F. Using Simple Sequence Repeats in 9 Brassica Complex Species to Assess Hypertrophic Curd Induction. Agriculture 2021, 11, 622. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, W.; Chu, P.; Wan, S.; Yang, M.; Wang, M.; Guan, R. Mapping a major QTL responsible for dwarf architecture in Brassica napus using a single-nucleotide polymorphism marker approach. BMC Plant Biol. 2016, 16, 178. [Google Scholar] [CrossRef] [PubMed]
- Treccarichi, S.; Ben Ammar, H.; Amari, M.; Cali, R.; Tribulato, A.; Branca, F. Molecular Markers for Detecting Inflorescence Size of Brassica oleracea L. Crops and B. oleracea Complex Species (n = 9) Useful for Breeding of Broccoli (B. oleracea var. italica) and Cauliflower (B. oleracea var. botrytis). Plants 2023, 12, 407. [Google Scholar] [CrossRef]
- Lv, H.; Wang, Q.; Liu, X.; Han, F.; Fang, Z.; Yang, L.; Zhuang, M.; Liu, Y.; Li, Z.; Zhang, Y. Whole-Genome Mapping Reveals Novel QTL Clusters Associated with Main Agronomic Traits of Cabbage (Brassica oleracea var. capitata L.). Front. Plant Sci. 2016, 7, 989. [Google Scholar] [CrossRef]
- Fridborg, I.; Kuusk, S.; Moritz, T.; Sundberg, E. The Arabidopsis dwarf mutant shi exhibits reduced gibberellin responses conferred by overexpression of a new putative zinc finger protein. Plant Cell 1999, 11, 1019–1032. [Google Scholar] [CrossRef]
- Youssef, H.M.; Eggert, K.; Koppolu, R.; Alqudah, A.M.; Poursarebani, N.; Fazeli, A.; Sakuma, S.; Tagiri, A.; Rutten, T.; Govind, G.; et al. VRS2 regulates hormone-mediated inflorescence patterning in barley. Nat. Genet. 2017, 49, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Yuo, T.; Yamashita, Y.; Kanamori, H.; Matsumoto, T.; Lundqvist, U.; Sato, K.; Ichii, M.; Jobling, S.A.; Taketa, S. A SHORT INTERNODES (SHI) family transcription factor gene regulates awn elongation and pistil morphology in barley. J. Exp. Bot. 2012, 63, 5223–5232. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Shi, P.; Lv, Y.; Gao, Z.; Chen, G. Gene coexpression network analysis reveals the role of SRS genes in senescence leaf of maize (Zea mays L.). J. Genet. 2020, 99, 3. [Google Scholar] [CrossRef] [PubMed]
- Bechtold, U.; Field, B. Molecular mechanisms controlling plant growth during abiotic stress. J. Exp. Bot. 2018, 69, 2753–2758. [Google Scholar] [CrossRef]
- Ben Ammar, H.; Picchi, V.; Arena, D.; Treccarichi, S.; Bianchi, G.; Lo Scalzo, R.; Marghali, S.; Branca, F. Variation of Bio-Morphometric Traits and Antioxidant Compounds of Brassica oleracea L. Accessions in Relation to Drought Stress. Agronomy 2022, 12, 2016. [Google Scholar] [CrossRef]
- Raza, A. Eco-physiological and Biochemical Responses of Rapeseed (Brassica napus L.) to Abiotic Stresses: Consequences and Mitigation Strategies. J. Plant Growth Regul. 2021, 40, 1368–1388. [Google Scholar] [CrossRef]
- Ma, L.; Wu, J.; Qi, W.; Coulter, J.A.; Fang, Y.; Li, X.; Liu, L.; Jin, J.; Niu, Z.; Yue, J.; et al. Screening and verification of reference genes for analysis of gene expression in winter rapeseed (Brassica rapa L.) under abiotic stress. PLoS ONE 2020, 15, e0236577. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, C.; Chen, H.; Wang, P.; Wang, P.; Song, C.; Zhang, X.; Wang, D. Multi-gene co-expression can improve comprehensive resistance to multiple abiotic stresses in Brassica napus L. Plant Sci. 2018, 274, 410–419. [Google Scholar] [CrossRef]
- Galletti, S.; Bagatta, M.; Branca, F.; Argento, S.; De Nicola, G.R.; Cianchetta, S.; Iori, R.; Ninfali, P. Isatis canescens is a rich source of glucobrassicin and other health-promoting compounds. J. Sci. Food Agric. 2015, 95, 158–164. [Google Scholar] [CrossRef]
- Büyük, İ.; Okay, A.; Aras, S. Identification and Characterization of SRS Genes in Phaseolus vulgaris Genome and Their Responses Under Salt Stress. Biochem. Genet. 2022, 60, 482–503. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent updates, new developments and status in 2020. Nucleic Acids Res. 2021, 49, D458–D460. [Google Scholar] [CrossRef]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Liang, F.; Gill, R.A.; Huang, J.; Cheng, X.; Liu, Y.; Tong, C.; Liu, S. A global survey of the transcriptome of allopolyploid Brassica napus based on single-molecule long-read isoform sequencing and Illumina-based RNA sequencing data. Plant J. 2020, 103, 843–857. [Google Scholar] [CrossRef]
- Zhang, Y.; Ali, U.; Zhang, G.; Yu, L.; Fang, S.; Iqbal, S.; Li, H.; Lu, S.; Guo, L. Transcriptome analysis reveals genes commonly responding to multiple abiotic stresses in rapeseed. Mol. Breed. 2019, 39, 158. [Google Scholar] [CrossRef]
- He, Y.; Li, Y.; Bai, Z.; Xie, M.; Zuo, R.; Liu, J.; Xia, J.; Cheng, X.; Liu, Y.; Tong, C.; et al. Genome-wide identification and functional analysis of cupin_1 domain-containing members involved in the responses to Sclerotinia sclerotiorum and abiotic stress in Brassica napus. Front. Plant Sci. 2022, 13, 983786. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Hu, M.; Xie, M.; Cui, X.; Huang, J.; Cheng, X.; Liu, L.; Liu, S.; Tong, C. Genome-Wide Characterization of Trehalose-6-Phosphate Synthase Gene Family of Brassica napus and Potential Links with Agronomic Traits. Int. J. Mol. Sci. 2022, 23, 15714. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool for Genome-wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef]
- Fang, D.; Zhang, W.; Cheng, X.; Hu, F.; Ye, Z.; Cao, J. Molecular evolutionary analysis of the SHI/STY gene family in land plants: A focus on the Brassica species. Front. Plant Sci. 2022, 13, 958964. [Google Scholar] [CrossRef]
- Zhang, Y.; von Behrens, I.; Zimmermann, R.; Ludwig, Y.; Hey, S.; Hochholdinger, F. LATERAL ROOT PRIMORDIA 1 of maize acts as a transcriptional activator in auxin signalling downstream of the Aux/IAA gene rootless with undetectable meristem 1. J. Exp. Bot. 2015, 66, 3855–3863. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Landherr, L.L.; Frohlich, M.W.; Leebens-Mack, J.; Ma, H.; dePamphilis, C.W. Patterns of gene duplication in the plant SKP1 gene family in angiosperms: Evidence for multiple mechanisms of rapid gene birth. Plant J. 2007, 50, 873–885. [Google Scholar] [CrossRef]
- Yang, Y.W.; Lai, K.N.; Tai, P.Y.; Ma, D.P.; Li, W.H. Molecular phylogenetic studies of Brassica, rorippa, arabidopsis and allied genera based on the internal transcribed spacer region of 18S-25S rDNA. Mol. Phylogenet. Evol. 1999, 13, 455–462. [Google Scholar] [CrossRef]
- Lysak, M.A.; Koch, M.A.; Pecinka, A.; Schubert, I. Chromosome triplication found across the tribe Brassiceae. Genome Res. 2005, 15, 516–525. [Google Scholar] [CrossRef]
- Huang, H.; Song, J.; Feng, Y.; Zheng, L.; Chen, Y.; Luo, K. Genome-Wide Identification and Expression Analysis of the SHI-Related Sequence Family in Cassava. Genes 2023, 14, 870. [Google Scholar] [CrossRef] [PubMed]
- Maire, P.; Wuarin, J.; Schibler, U. The role of cis-acting promoter elements in tissue-specific albumin gene expression. Science 1989, 244, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, A.; Yamamoto, M. Cis-element architecture of Nrf2-sMaf heterodimer binding sites and its relation to diseases. Arch. Pharm. Res. 2020, 43, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Oudelaar, A.M.; Higgs, D.R. The relationship between genome structure and function. Nat. Rev. Genet. 2021, 22, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.T.; Xu, H.H.; Li, J.; Lu, Y.T. Auxin abolishes SHI-RELATED SEQUENCE5-mediated inhibition of lateral root development in Arabidopsis. New Phytol. 2020, 225, 297–309. [Google Scholar] [CrossRef]
- Lütken, H.; Jensen, L.S.; Topp, S.H.; Mibus, H.; Müller, R.; Rasmussen, S.K. Production of compact plants by overexpression of AtSHI in the ornamental Kalanchoë. Plant Biotechnol. J. 2010, 8, 211–222. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, M.; Xie, M.; Cui, X.; Huang, J.; Cheng, X.; Liu, L.; Yan, S.; Liu, S.; Tong, C. Characterization and Potential Function Analysis of the SRS Gene Family in Brassica napus. Genes 2023, 14, 1421. https://doi.org/10.3390/genes14071421
Hu M, Xie M, Cui X, Huang J, Cheng X, Liu L, Yan S, Liu S, Tong C. Characterization and Potential Function Analysis of the SRS Gene Family in Brassica napus. Genes. 2023; 14(7):1421. https://doi.org/10.3390/genes14071421
Chicago/Turabian StyleHu, Ming, Meili Xie, Xiaobo Cui, Junyan Huang, Xiaohui Cheng, Lijiang Liu, Shunping Yan, Shengyi Liu, and Chaobo Tong. 2023. "Characterization and Potential Function Analysis of the SRS Gene Family in Brassica napus" Genes 14, no. 7: 1421. https://doi.org/10.3390/genes14071421
APA StyleHu, M., Xie, M., Cui, X., Huang, J., Cheng, X., Liu, L., Yan, S., Liu, S., & Tong, C. (2023). Characterization and Potential Function Analysis of the SRS Gene Family in Brassica napus. Genes, 14(7), 1421. https://doi.org/10.3390/genes14071421