
Gene Genealogy-Based Mutation Analysis Reveals Emergence of Aus, Tropical japonica, and Aromatic of Oryza sativa during the Later Stage of Rice Domestication

Abstract
1. Introduction
2. Materials and Methods
2.1. Study Samples
2.2. Gene Genealogy-Based Reconstruction of Intra-Specific Phylogeny
2.2.1. Classification of Mutations Based on Gene Genealogies
2.2.2. Gene Genealogies Sampled for Phylogenetic Reconstruction
2.2.3. Inference of Phylogeny from Distributions of Mutations among Subgroup Combinations
3. Results
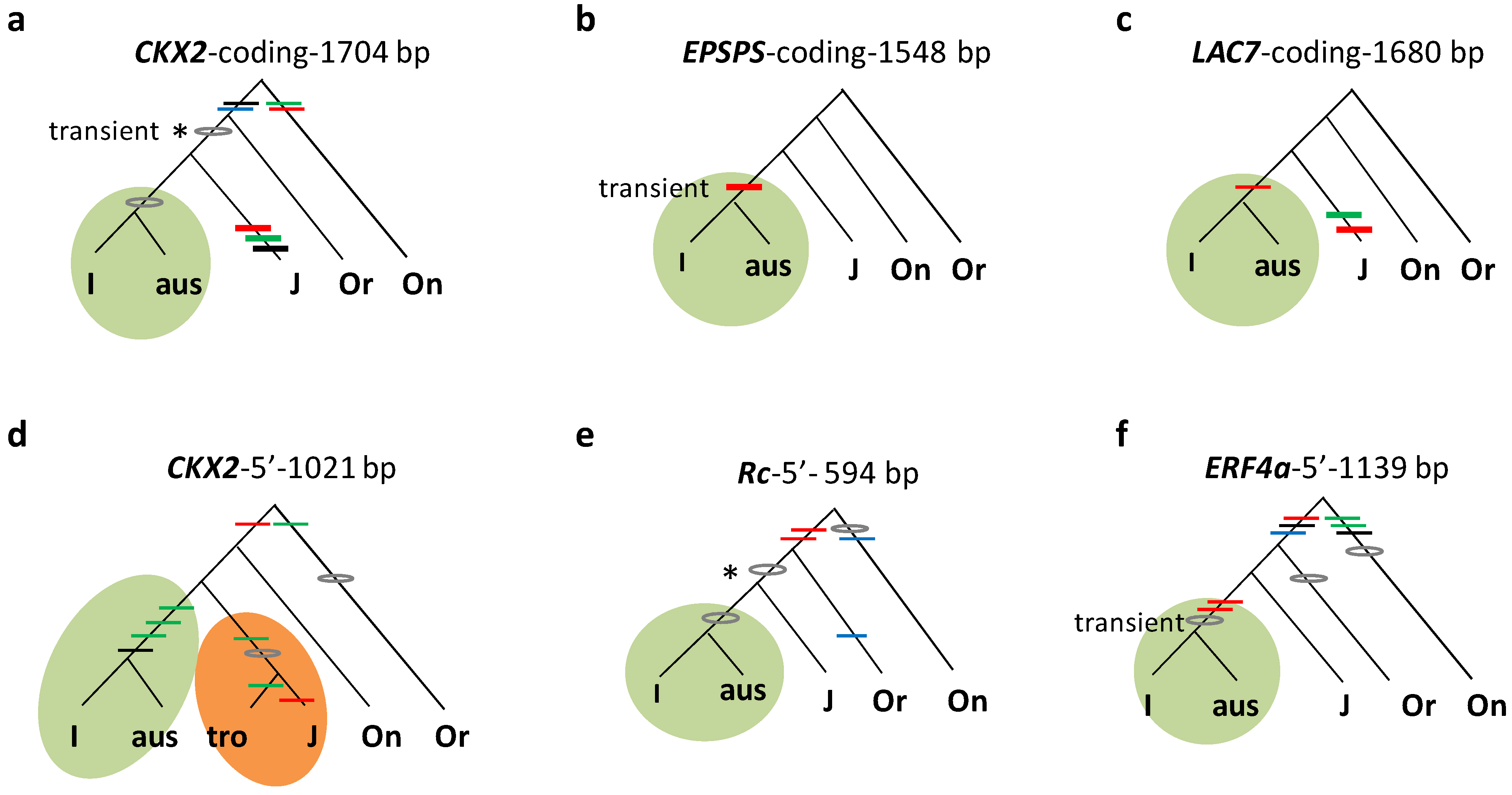
3.1. All Five Subgroups Share the Same Early Mutations
3.2. Subgroup Aus Differentiated in Indica and Tropical Japonica in Japonica
3.3. Subgroup Aromatic Was Derived from Hybrid Progeny between Aus and Tropical Japonica
3.4. Subgroup Aus Differentiated from Indica Earlier than Tropical Japonica from Japonica
3.5. Phylogeny Reconstructed from Collective Evidence
3.6. Validity of Mutation Identification
4. Discussion
4.1. GGM-Based Analysis and Specific Test for Asian Rice
4.2. The Early Rice Laid the Foundation for All Subgroups of Asian Rice
4.3. Subgroup Aus Branched off Early in Indica
4.4. Aromatic Rice as Hybrid Progeny of Aus and Tropical japonica
4.5. Applications and Error Rates
4.6. Implications of the Phylogeny
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baack, E.J.; Rieseberg, L.H. A genomic view of introgression and hybrid speciation. Curr. Opin. Genet. Dev. 2007, 17, 513–518. [Google Scholar] [CrossRef]
- Lu, Y.Q.; Xu, Y.Z.; Li, N. Early domestication history of Asian rice revealed by mutations and genome-wide analysis of gene genealogies. Rice 2022, 15, 20. [Google Scholar] [CrossRef] [PubMed]
- Glaszmann, J.C. Isozymes and classification of Asian rice varieties. Theor. Appl. Genet. 1987, 74, 21–30. [Google Scholar] [CrossRef]
- Parsons, B.J.; Newbury, H.J.; Jackson, M.T.; Ford-Lloyd, B.V. The genetic structure and conservation of aus, aman and boro rices from Bangladesh. Genet. Resour. Crop Evol. 1999, 46, 587–598. [Google Scholar] [CrossRef]
- Garris, A.J.; Tai, T.H.; Coburn, J.; Kresovich, S.; McCouch, S. Genetic structure and diversity in Oryza sativa L. Genetics 2005, 169, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.J. The Middle Yangtze region in China is one place where rice was domesticated: Phytolith evidence from the Diaotonghuan Cave, Northern Jiangxi. Antiquity 1998, 72, 885–897. [Google Scholar]
- Gross, B.L.; Zhao, Z.J. Archaeological and genetic insights into the origins of domesticated rice. Proc. Natl. Acad. Sci. USA 2014, 111, 6190–6197. [Google Scholar] [CrossRef] [PubMed]
- Khush, G.S. Origin, dispersal, cultivation and variation of rice. Plant Mol.Biol. 1997, 35, 25–34. [Google Scholar] [CrossRef]
- Ali, M.L.; McClung, A.M.; Jia, M.H.; Kimball, J.A.; McCouch, S.R.; Eizenga, G.C. A rice diversity panel evaluated for genetic and agro-morphological diversity between subpopulations and its geographic distribution. Crop Sci. 2011, 51, 2021–2035. [Google Scholar]
- Schatz, M.C.; Maron, L.G.; Stein, J.C.; Hernandez Wences, A.; Gurtowski, J.; Biggers, E.; Lee, H.; Kramer, M.; Antoniou, E.; Ghiban, E.; et al. Whole genome de novo assemblies of three divergent strains of rice, Oryza sativa, document novel gene space of aus and indica. Genome Biol. 2014, 15, 506. [Google Scholar]
- Wang, W.S.; Mauleon, R.; Hu, Z.Q.; Chebotarov, D.; Tai, S.S.; Wu, Z.C.; Li, M.; Zheng, T.Q.; Fuentes, R.R.; Zhang, F.; et al. Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature 2018, 557, 43–49. [Google Scholar] [CrossRef]
- Mackill, D.J.; Lei, X.M. Genetic variation for traits related to temperate adaptation of rice cultivars. Crop Sci. 1997, 37, 1340–1346. [Google Scholar] [CrossRef]
- Carpentier, M.C.; Manfroi, E.; Wei, F.J.; Wu, H.P.; Lasserre, E.; Llauro, C.; Debladis, E.; Akakpo, R.; Hsing, Y.I.; Panaud, O. Retrotranspositional landscape of Asian rice revealed by 3000 genomes. Nat. Commun. 2019, 10, 12. [Google Scholar] [CrossRef]
- Chin, J.H.; Gamuyao, R.; Dalid, C.; Bustamam, M.; Prasetiyono, J.; Moeljopawiro, S.; Wissuwa, M.; Heuer, S. Developing rice with high yield under phosphorus deficiency: Pup1 sequence to application. Plant Physiol. 2011, 156, 1202–1216. [Google Scholar] [CrossRef]
- Gamuyao, R.; Chin, J.H.; Pariasca-Tanaka, J.; Pesaresi, P.; Catausan, S.; Dalid, C.; Slamet-Loedin, I.; Tecson-Mendoza, E.M.; Wissuwa, M.; Heuer, S. The protein kinase Pstol1 from traditional rice confers tolerance of phosphorus deficiency. Nature 2012, 488, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.J.; Wang, L.; Pan, Q.H. A new recessive gene conferring resistance against rice blast. Rice 2016, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Casartelli, A.; Riewe, D. Exploring traditional aus-type rice for metabolites conferring drought tolerance. Rice 2018, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Civan, P.; Craig, H.; Cox, C.J.; Brown, T.A. Three geographically separate domestications of Asian rice. Nat. Plants 2015, 1, 15164. [Google Scholar] [CrossRef]
- Santos, J.D.; Chebotarov, D.; McNally, K.L.; Bartholomé, J.; Droc, G.; Billot, C.; Glaszmann, J.C. Fine scale genomic signals of admixture and alien introgression among Asian rice landraces. Genome Biol. Evol. 2019, 11, 1358–1373. [Google Scholar] [CrossRef]
- Civáň, P.; Ali, S.; Batista-Navarro, R.; Drosou, K.; Ihejieto, C.; Chakraborty, D.; Ray, A.; Gladieux, P.; Brown, T.A. Origin of the aromatic group of cultivated rice (Oryza sativa L.) traced to the Indian subcontinent. Genome Biol. Evol. 2019, 11, 832–843. [Google Scholar] [CrossRef]
- Choi, J.Y.; Lye, Z.N.; Groen, S.C.; Dai, X.G.; Rughani, P.; Zaaijer, S.; Harrington, E.D.; Juul, S.; Purugganan, M.D. Nanopore sequencing-based genome assembly and evolutionary genomics of circum-basmati rice. Genome Biol. 2020, 21, 27. [Google Scholar] [CrossRef] [PubMed]
- Maga, J.A. Rice product volatiles—A review. J. Agric. Food Chem. 1984, 32, 964–970. [Google Scholar] [CrossRef]
- Widjaja, R.; Craske, J.D.; Wootton, M. Comparative studies on volatile components of non-fragrant and fragrant rices. J. Sci. Food Agric. 1996, 70, 151–161. [Google Scholar] [CrossRef]
- Buttery, R.G.; Ling, L.C.; Juliano, B.O.; Turnbaugh, J.G. Cooked rice aroma and 2-acetyl-1-pyrroline. J. Agric. Food Chem. 1983, 31, 823–826. [Google Scholar] [CrossRef]
- Wakte, K.; Zanan, R.; Hinge, V.; Khandagale, K.; Nadaf, A.; Henry, R. Thirty-three years of 2-acetyl-1-pyrroline, a principal basmati aroma compound in scented rice (Oryza sativa L.): A status review. J. Sci. Food Agric. 2017, 97, 384–395. [Google Scholar] [CrossRef]
- Yoshihashi, T.; Huong, N.T.T.; Inatomi, H. Precursors of 2-acetyl-1-pyrroline, a potent flavor compound of an aromatic rice variety. J. Agric. Food Chem. 2002, 50, 2001–2004. [Google Scholar] [CrossRef]
- Bradbury, L.M.T.; Fitzgerald, T.L.; Henry, R.J.; Jin, Q.S.; Waters, D.L.E. The gene for fragrance in rice. Plant Biotechnol. J. 2007, 3, 363–370. [Google Scholar] [CrossRef]
- Chen, S.H.; Yang, Y.; Shi, W.W.; Ji, Q.; He, F.; Zhang, Z.D.; Cheng, Z.K.; Liu, X.N.; Xu, M.L. Badh2, encoding betaine aldehyde dehydrogenase, inhibits the biosynthesis of 2-acetyl-1-pyrroline, a major component in rice fragrance. Plant Cell 2008, 20, 1850–1861. [Google Scholar] [CrossRef]
- Singh, A.; Singh, P.K.; Singh, R.; Pandit, A.; Mahato, A.K.; Gupta, D.K.; Tyagi, K.; Singh, A.K.; Singh, N.K.; Sharma, T.R. SNP haplotypes of the BADH1 gene and their association with aroma in rice (Oryza sativa L.). Mol. Breed. 2010, 26, 325–338. [Google Scholar] [CrossRef]
- He, Q.; Yu, J.; Kim, T.S.; Cho, Y.H.; Lee, Y.S.; Park, Y.J. Resequencing reveals different domestication rate for BADH1 and BADH2 in rice (Oryza sativa). PLoS ONE 2015, 10, 12. [Google Scholar] [CrossRef]
- Bourgis, F.; Guyot, R.; Gherbi, H.; Tailliez, E.; Amabile, I.; Salse, J.; Lorieux, M.; Delseny, M.; Ghesquiere, A. Characterization of the major fragance gene from an aromatic japonica rice and analysis of its diversity in Asian cultivated rice. Theor. Appl. Genet. 2008, 117, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Kovach, M.J.; Calingacion, M.N.; Fitzgerald, M.A.; McCouch, S.R. The origin and evolution of fragrance in rice (Oryza sativa L.). Proc. Natl. Acad. Sci. USA 2009, 106, 14444–14449. [Google Scholar] [CrossRef]
- Sakthivel, K.; Sundaram, R.M.; Rani, N.S.; Balachandran, S.M.; Neeraja, C.N. Genetic and molecular basis of fragrance in rice. Biotechnol. Adv. 2009, 27, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Myint, K.M.; Courtois, B.; Risterucci, A.M.; Frouin, J.; Soe, K.; Thet, K.M.; Vanavichit, A.; Glaszmann, J.C. Specific patterns of genetic diversity among aromatic rice varieties in Myanmar. Rice 2012, 5, 20. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.W.; Li, W.; Pan, S.G.; Fitzgerald, T.L.; Xiao, F.; Tang, Y.J.; Wang, Y.L.; Duan, M.Y.; Tian, H.; Tang, X.R. Shading during the grain filling period increases 2-acetyl-1-pyrroline content in fragrant rice. Rice 2015, 8, 10. [Google Scholar] [CrossRef]
- Gay, F.; Maraval, I.; Roques, S.; Gunata, Z.; Boulanger, R.; Audebert, A.; Mestres, C. Effect of salinity on yield and 2-acetyl-1-pyrroline content in the grains of three fragrant rice cultivars (Oryza sativa L.) in Camargue (France). Field Crop. Res. 2010, 117, 154–160. [Google Scholar] [CrossRef]
- Banerjee, A.; Ghosh, P.; Roychoudhury, A. Salt acclimation differentially regulates the metabolites commonly involved in stress tolerance and aroma synthesis in indica rice cultivars. Plant Growth Regul. 2019, 88, 87–97. [Google Scholar] [CrossRef]
- Li, W.-H. Molecular Evolution; Sinauer Associates: Sunderland, MA, USA, 1997. [Google Scholar]
- Nordborg, M.; Donnelly, P. The coalescent process with selfing. Genetics 1997, 146, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.P.; Zhang, J.Z. The pleiotropic structure of the genotype-phenotype map: The evolvability of complex organisms. Nat. Rev. Genet. 2011, 12, 204–213. [Google Scholar] [CrossRef]
- Shivrain, V.K.; Burgos, N.R.; Anders, M.M.; Rajguru, S.N.; Moore, J.; Sales, M.A. Gene flow between Clearfield (TM) rice and red rice. Crop Prot. 2007, 26, 349–356. [Google Scholar] [CrossRef]
- Long, T.W.; Chen, H.S.; Leipe, C.; Wagner, M.; Tarasov, P.E. Modelling the chronology and dynamics of the spread of Asian rice from ca. 8000 BCE to 1000 CE. Quat. Int. 2022, 623, 101–109. [Google Scholar] [CrossRef]
- Gutaker, R.M.; Groen, S.C.; Bellis, E.S.; Choi, J.Y.; Pires, I.S.; Bocinsky, R.K.; Slayton, E.R.; Wilkins, O.; Castillo, C.C.; Negrao, S.; et al. Genomic history and ecology of the geographic spread of rice. Nat. Plants 2020, 6, 492–502. [Google Scholar] [CrossRef]
- Deng, Z.H.; Huang, B.X.; Zhang, Q.L.; Zhang, M. First farmers in the south China coast: New evidence from the Gancaoling site of Guangdong province. Front. Earth Sci. 2022, 10, 11. [Google Scholar] [CrossRef]
- Wang, W.W.; Nguyen, K.D.; Le, H.D.; Zhao, C.G.; Carson, M.T.; Yang, X.Y.; Hung, H.C. Before rice and the first rice: Archaeobotanical study in Ha Long Bay, Northern Vietnam. Front. Earth Sci. 2022, 10, 15. [Google Scholar]
- Wang, C.H.; Zheng, X.M.; Xu, Q.; Yuan, X.P.; Huang, L.; Zhou, H.F.; Wei, X.H.; Ge, S. Genetic diversity and classification of Oryza sativa with emphasis on Chinese rice germplasm. Heredity 2014, 112, 489–496. [Google Scholar] [CrossRef]
- Chang, T.T. Origin, evolution, cultivation, dissemination, and diversification of Asian and African rices. Euphytica 1976, 25, 425–441. [Google Scholar] [CrossRef]
- Bligh, H.F.J. Detection of adulteration of Basmati rice with non-premium long-grain rice. Int. J. Food Sci. Technol. 2000, 35, 257–265. [Google Scholar] [CrossRef]
- Bhattacharjee, P.; Singhal, R.S.; Kulkarni, P.R. Basmati rice: A review. Int. J. Food Sci. Technol. 2002, 37, 1–12. [Google Scholar] [CrossRef]
- Londo, J.P.; Chiang, Y.C.; Hung, K.H.; Chiang, T.Y.; Schaal, B.A. Phylogeography of Asian wild rice, Oryza rufipogon, reveals multiple independent domestications of cultivated rice, Oryza sativa. Proc. Natl. Acad. Sci. USA 2006, 103, 9578–9583. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.M.; Ge, S. Ecological divergence in the presence of gene flow in two closely related Oryza species (Oryza rufipogon and O. nivara). Mol. Ecol. 2010, 19, 2439–2454. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Platts, A.E.; Fuller, D.Q.; Hsing, Y.-I.; Wing, R.A.; Purugganan, M.D. The rice paradox: Multiple origins but single domestication in Asian rice. Mol. Biol. Evol. 2017, 34, 969–979. [Google Scholar] [CrossRef]
- Cortés, A.J.; Chavarro, M.C.; Madrinan, S.; This, D.; Blair, M.W. Molecular ecology and selection in the drought-related Asr gene polymorphisms in wild and cultivated common bean (Phaseolus vulgaris L.). BMC Genet. 2012, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, N.; Ramanantsoanirina, A.; Santos, J.D.; Frouin, J.; Radanielina, T. Evolutionary processes involved in the emergence and expansion of an atypical O. sativa group in Madagascar. Rice 2021, 14, 44. [Google Scholar] [CrossRef] [PubMed]
- Suvi, W.T.; Shimelis, H.; Laing, M. Breeding rice for rice yellow mottle virus resistance in Sub-Saharan Africa: A review. Acta Agric. Scand. Sect. B-Soil Plant Sci. 2019, 69, 181–188. [Google Scholar] [CrossRef]
- Cruz, M.; Arbelaez, J.D.; Loaiza, K.; Cuasquer, J.; Rosas, J.; Graterol, E. Genetic and phenotypic characterization of rice grain quality traits to define research strategies for improving rice milling, appearance, and cooking qualities in Latin America and the Caribbean. Plant Genome 2021, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Cortés, A.J.; López-Hernández, F. Harnessing crop wild diversity for climate change adaptation. Genes 2021, 12, 783. [Google Scholar] [CrossRef]
- Ashikari, M.; Sakakibara, H.; Lin, S.Y.; Yamamoto, T.; Takashi, T.; Nishimura, A.; Angeles, E.R.; Qian, Q.; Kitano, H.; Matsuoka, M. Cytokinin oxidase regulates rice grain production. Science 2005, 309, 741–745. [Google Scholar] [CrossRef]
- Furukawa, T.; Maekawa, M.; Oki, T.; Suda, I.; Iida, S.; Shimada, H.; Takamure, I.; Kadowaki, K.I. The Rc and Rd genes are involved in proanthocyanidin synthesis in rice pericarp. Plant J. 2007, 49, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Huo, X.; Wu, S.; Zhu, Z.F.; Liu, F.X.; Fu, Y.C.; Cai, H.W.; Sun, X.Y.; Gu, P.; Xie, D.X.; Tan, L.B.; et al. NOG1 increases grain production in rice. Nat. Commun. 2017, 8, 11. [Google Scholar] [CrossRef]
- Joo, J.; Choi, H.J.; Lee, Y.H.; Kim, Y.K.; Song, S.I. A transcriptional repressor of the ERF family confers drought tolerance to rice and regulates genes preferentially located on chromosome 11. Planta 2013, 238, 155–170. [Google Scholar] [CrossRef]
- Swetha, C.; Basu, D.; Pachamuthu, K.; Tirumalai, V.; Nair, A.; Prasad, M.; Shivaprasad, P.V. Major domestication-related phenotypes in Indica rice are due to loss of miRNA-mediated laccase silencing. Plant Cell 2018, 30, 2649–2662. [Google Scholar] [CrossRef]
- Ashikari, M.; Sasaki, A.; Ueguchi-Tanaka, M.; Itoh, H.; Nishimura, A.; Datta, S.; Ishiyama, K.; Saito, T.; Kobayashi, M.; Khush, G.S.; et al. Loss-of-function of a rice gibberellin biosynthetic gene, GA20 oxidase (GA20ox-2), led to the rice ’green revolution’. Breed. Sci. 2002, 52, 143–150. [Google Scholar] [CrossRef]
- Chen, J.; Gao, H.; Zheng, X.M.; Jin, M.N.; Weng, J.F.; Ma, J.; Ren, Y.L.; Zhou, K.N.; Wang, Q.; Wang, J.; et al. An evolutionarily conserved gene, FUWA, plays a role in determining panicle architecture, grain shape and grain weight in rice. Plant J. 2015, 83, 427–438. [Google Scholar] [CrossRef]
- Song, X.J.; Huang, W.; Shi, M.; Zhu, M.Z.; Lin, H.X. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nature Genet. 2007, 39, 623–630. [Google Scholar] [CrossRef]
- Wu, C.Y.; Trieu, A.; Radhakrishnan, P.; Kwok, S.F.; Harris, S.; Zhang, K.; Wang, J.L.; Wan, J.M.; Zhai, H.Q.; Takatsuto, S.; et al. Brassinosteroids regulate grain filling in rice. Plant Cell 2008, 20, 2130–2145. [Google Scholar] [CrossRef]
- Zheng, J.; Wu, H.; Zhao, M.C.; Yang, Z.A.; Zhou, Z.H.; Guo, Y.M.; Lin, Y.J.; Chen, H. OsMYB3 is a R2R3-MYB gene responsible for anthocyanin biosynthesis in black rice. Mol. Breed. 2021, 41, 15. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Fang, J.; Ou, S.J.; Gao, S.P.; Zhang, F.X.; Du, L.; Xiao, Y.H.; Wang, H.R.; Sun, X.H.; Chu, J.F.; et al. Variations in CYP78A13 coding region influence grain size and yield in rice. Plant Cell Environ. 2015, 38, 800–811. [Google Scholar] [CrossRef]
- Dong, H.J.; Zhao, H.; Xie, W.B.; Han, Z.M.; Li, G.W.; Yao, W.; Bai, X.F.; Hu, Y.; Guo, Z.L.; Lu, K.; et al. A novel tiller angle gene, TAC3, together with TAC1 and D2 largely determine the natural variation of tiller angle in rice cultivars. PLoS Genet. 2016, 12. [Google Scholar] [CrossRef]
- Takahashi, Y.; Shomura, A.; Sasaki, T.; Yano, M. Hd6, a rice quantitative trait locus involved in photoperiod sensitivity, encodes the alpha subunit of protein kinase CK2. Proc. Natl. Acad. Sci. USA 2001, 98, 7922–7927. [Google Scholar] [CrossRef]
- Li, S.Y.; Zhao, B.R.; Yuan, D.Y.; Duan, M.J.; Qian, Q.; Tang, L.; Wang, B.; Liu, X.Q.; Zhang, J.; Wang, J.; et al. Rice zinc finger protein DST enhances grain production through controlling Gn1a/OsCKX2 expression. Proc. Natl. Acad. Sci. USA 2013, 110, 3167–3172. [Google Scholar] [CrossRef]
- Shih, C.H.; Chu, H.; Tang, L.K.; Sakamoto, W.; Maekawa, M.; Chu, I.K.; Wang, M.; Lo, C. Functional characterization of key structural genes in rice flavonoid biosynthesis. Planta 2008, 228, 1043–1054. [Google Scholar] [CrossRef]
- Wang, E.; Wang, J.; Zhu, X.D.; Hao, W.; Wang, L.Y.; Li, Q.; Zhang, L.X.; He, W.; Lu, B.R.; Lin, H.X.; et al. Control of rice grain-filling and yield by a gene with a potential signature of domestication. Nature Genet. 2008, 40, 1370–1374. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Yun, K.Y.; Ressom, H.W.; Mohanty, B.; Bajic, V.B.; Jia, Y.L.; Yun, S.J.; de los Reyes, B.G. An early response regulatory cluster induced by low temperature and hydrogen peroxide in seedlings of chilling-tolerant japonica rice. BMC Genomics 2007, 8, 18. [Google Scholar] [CrossRef]
- Gu, B.G.; Zhou, T.Y.; Luo, J.H.; Liu, H.; Wang, Y.C.; Shangguan, Y.Y.; Zhu, J.J.; Li, Y.; Sang, T.; Wang, Z.X.; et al. An-2 encodes a cytokinin synthesis enzyme that regulates awn length and grain production in rice. Mol. Plant. 2015, 8, 1635–1650. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.C.; Xiao, Y.H.; Niu, M.; Meng, W.J.; Li, L.L.; Zhang, X.X.; Liu, D.P.; Zhang, G.X.; Qian, Y.W.; Sun, Z.T.; et al. ARGONAUTE2 enhances grain length and salt tolerance by activating BIG GRAIN3 to modulate cytokinin distribution in rice. Plant Cell 2020, 32, 2292–2306. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Tanaka, K.; Kuwano, M.; Yoshida, K.T. Expression pattern of inositol phosphate-related enzymes in rice (Oryza sativa L.): Implications for the phytic acid biosynthetic pathway. Gene 2007, 405, 55–64. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, Y.J.; Kim, B.G.; Lim, Y.; Ahn, J.H. Flavanone 3 beta-hydroxylases from rice: Key enzymes for favonol and anthocyanin biosynthesis. Mol. Cells 2008, 25, 312–316. [Google Scholar]
- Li, C.B.; Zhou, A.L.; Sang, T. Rice domestication by reducing shattering. Science 2006, 311, 1936–1939. [Google Scholar] [CrossRef]
- Li, Y.B.; Fan, C.C.; Xing, Y.Z.; Jiang, Y.H.; Luo, L.J.; Sun, L.; Shao, D.; Xu, C.J.; Li, X.H.; Xiao, J.H.; et al. Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nature Genet. 2011, 43, 1266–1269. [Google Scholar] [CrossRef]
- Iwai, T.; Miyasaka, A.; Seo, S.; Ohashi, Y. Contribution of ethylene biosynthesis for resistance to blast fungus infection in young rice plants. Plant Physiol. 2006, 142, 1202–1215. [Google Scholar] [CrossRef]
- Yoon, J.; Cho, L.H.; Kim, S.L.; Choi, H.; Koh, H.J.; An, G. The BEL1-type homeobox gene SH5 induces seed shattering by enhancing abscission-zone development and inhibiting lignin biosynthesis. Plant J. 2014, 79, 717–728. [Google Scholar] [CrossRef]
- Li, H.W.; Zang, B.S.; Deng, X.W.; Wang, X.P. Overexpression of the trehalose-6-phosphate synthase gene OsTPS1 enhances abiotic stress tolerance in rice. Planta 2011, 234, 1007–1018. [Google Scholar] [CrossRef]
- Xu, J.W.; Feng, D.J.; Li, X.G.; Chang, T.J.; Zhu, Z. Cloning of genomic DNA of rice 5-enolpyruvylshikimate 3-phosphate synthase gene and chromosomal localization of the gene. Sci. China Ser. C-Life Sci. 2002, 45, 251–259. [Google Scholar] [CrossRef]
- Kojima, S.; Takahashi, Y.; Kobayashi, Y.; Monna, L.; Sasaki, T.; Araki, T.; Yano, M. Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell Physiol. 2002, 43, 1096–1105. [Google Scholar] [CrossRef]
- Reddy, V.S.; Scheffler, B.E.; Wienand, U.; Wessler, S.R.; Reddy, A.R. EMBL accession #: Y15219. Plant Mol.Biol. 1998, 36, 497–498. [Google Scholar]
- Liu, Y.; Wang, H.; Jiang, Z.; Wang, W.; Xu, R.; Wang, Q.; Zhang, Z.; Li, A.; Liang, Y.; Ou, S.; et al. Genomic basis of geographical adaptation to soil nitrogen in rice. Nature 2021, 590, 600–605. [Google Scholar] [CrossRef]
- Yano, M.; Katayose, Y.; Ashikari, M.; Yamanouchi, U.; Monna, L.; Fuse, T.; Baba, T.; Yamamoto, K.; Umehara, Y.; Nagamura, Y.; et al. Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 2000, 12, 2473–2483. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Huang, W.; Gao, J.P.; Yang, J.; Shi, M.; Zhu, M.Z.; Luo, D.; Lin, H.X. Genetic control of rice plant architecture under domestication. Nature Genet. 2008, 40, 1365–1369. [Google Scholar] [CrossRef]
- Sweeney, M.T.; Thomson, M.J.; Pfeil, B.E.; McCouch, S. Caught red-handed: Rc encodes a basic helix-loop-helix protein conditioning red pericarp in rice. Plant Cell 2006, 18, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Si, L.Z.; Chen, J.Y.; Huang, X.H.; Gong, H.; Luo, J.H.; Hou, Q.Q.; Zhou, T.Y.; Lu, T.T.; Zhu, J.J.; Shangguan, Y.Y.; et al. OsSPL13 controls grain size in cultivated rice. Nature Genet. 2016, 48, 447–456. [Google Scholar] [CrossRef]
- Huang, Y.; Bai, X.F.; Cheng, N.N.; Xiao, J.H.; Li, X.H.; Xing, Y.Z. Wide Grain 7 increases grain width by enhancing H3K4me3 enrichment in the OsMADS1 promoter in rice (Oryza sativa L.). Plant J. 2020, 102, 517–528. [Google Scholar] [CrossRef]
- Bessho-Uehara, K.; Wang, D.R.; Furuta, T.; Minami, A.; Nagai, K.; Gamuyao, R.; Asano, K.; Angeles-Shim, R.B.; Shimizu, Y.; Ayano, M.; et al. Loss of function at RAE2, a previously unidentified EPFL, is required for awnlessness in cultivated Asian rice. Proc. Natl. Acad. Sci. USA 2016, 113, 8969–8974. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.K.; Li, S.; Liu, Q.; Wu, K.; Zhang, J.Q.; Wang, S.S.; Wang, Y.; Chen, X.B.; Zhang, Y.; Gao, C.X.; et al. The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nature Genet. 2015, 47, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.Z.; Qian, Q.; Liu, Z.B.; Sun, H.Y.; He, S.Y.; Luo, D.; Xia, G.M.; Chu, C.C.; Li, J.Y.; Fu, X.D. Natural variation at the DEP1 locus enhances grain yield in rice. Nature Genet. 2009, 41, 494–497. [Google Scholar] [CrossRef]
- Jung, K.H.; Lee, J.; Dardick, C.; Seo, Y.S.; Cao, P.; Canlas, P.; Phetsom, J.; Xu, X.; Ouyang, S.; An, K.; et al. Identification and functional analysis of light-responsive unique genes and gene family members in rice. PLoS Genet. 2008, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Ashikari, M.; Miura, K.; Yamashino, T.; Mizuno, T. The evolutionarily conserved OsPRR quintet: Rice pseudo-response regulators implicated in circadian rhythm. Plant Cell Physiol. 2003, 44, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Tohge, T.; Watanabe, M.; Hoefgen, R.; Fernie, A.R. Shikimate and phenylalanine biosynthesis in the green lineage. Front. Plant Sci. 2013, 4, 13. [Google Scholar] [CrossRef]
- Cai, Q.; Yuan, Z.; Chen, M.J.; Yin, C.S.; Luo, Z.J.; Zhao, X.X.; Liang, W.Q.; Hu, J.P.; Zhang, D.B. Jasmonic acid regulates spikelet development in rice. Nat. Commun. 2014, 5, 13. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Gao, P.; Xing, Z.; Jin, S.M.; Chen, Z.D.; Liu, L.T.; Constantino, N.; Wang, X.W.; Shi, W.B.; Yuan, J.S.; et al. Application of an improved proteomics method for abundant protein cleanup: Molecular and genomic mechanisms study in plant defense. Mol. Cell. Proteom. 2013, 12, 3431–3442. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Taylor, J.M.; Spriggs, A.; Zhang, H.Y.; Wu, X.J.; Russell, S.; Singh, M.; Koltunow, A. A genome-wide survey of imprinted genes in rice seeds reveals imprinting primarily occurs in the endosperm. PLoS Genet. 2011, 7, 14. [Google Scholar] [CrossRef]
- Reddy, A.R.; Scheffler, B.; Madhuri, G.; Srivastava, M.N.; Kumar, A.; Sathyanarayanan, P.V.; Nair, S.; Mohan, M. Chalcone synthase in rice (Oryza sativa L.): Detection of the CHS protein in seedlings and molecular mapping of the chs locus. Plant Mol. Biol. 1996, 32, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Bryan, G.T.; Wu, K.S.; Farrall, L.; Jia, Y.L.; Hershey, H.P.; McAdams, S.A.; Faulk, K.N.; Donaldson, G.K.; Tarchini, R.; Valent, B. A single amino acid difference distinguishes resistant and susceptible alleles of the rice blast resistance gene Pi-ta. Plant Cell 2000, 12, 2033–2045. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Chromosome | Gene a | Length (bp) b | Early Mutations c | Mutation Density d | Mutations Specific to Two Subgroups | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aus and indica e | Mutation Density | tropical japonica and japonica f | Mutation Density g | |||||||||
| 5′ | Coding | 5′ | Coding | 5′ | Coding | 5′ | Coding | |||||
| 1 | CKX2 | 1021 | 1704 | 0 (5′), 1 (coding) | 4 (5′), 1 (coding) | 0.0039 | 0.0006 | 2 (5′), 0 (coding) f | 0.0020 | |||
| DFR | 1036 | 1119 | 1 (5′), 1 (5′), 0 (coding) | 0.0010 | ||||||||
| NOG1 | 1012 | 1170 | 20 (5′), 1 (coding) f | 0.0198 | 0.0009 | |||||||
| ERF4a a | 1090 | 708 | 3 (5′), 1 (coding) | 0.0028 | 0.0014 | |||||||
| LAC7 a | 1025 | 1680 | 0 (5′), 1 (coding) | 0.0006 | 2 (5′), 2 (coding) f | 0.0020 | 0.0012 | |||||
| SD1 | 1004 | 1170 | 0 (5′), 1 (coding) | 0.0009 | 0 (5′), 1 (coding) | 0.0009 | 4 (5′), 2 (coding) f | 0.0040 | 0.0017 | |||
| 2 | FUWA a | 1028 | 1482 | 1 (5′), 0 (coding) e | 0.0010 | 10 (5′), 0 (coding) | 0.0097 | |||||
| GW2 a | 1020 | 1278 | 8 (5′), 0 (coding) | 0.0078 | ||||||||
| SK2 | 1013 | 1029 | 1 (5′), 1 (5′), 0 (coding) | 0.0010 | 7 (5′), 1 (coding) f | 0.0069 | 0.0010 | |||||
| 3 | SUS4 | 1057 | 2430 | 4 (5′), 1 (coding) f | 0.0038 | 0.0004 | ||||||
| MYB3 | 1017 | 1053 | 0 (5′), 2 (coding) | |||||||||
| GL3.2 | 1153 | 1554 | 1 (5′), 3 (5′), 3 (coding) | 0.0009 | 1 (5′), 0 (coding) | 0.0009 | ||||||
| TAC3 a | 1024 | 459 | 1 (5′), 0 (coding) e | 0.0010 | 1 (5′), 0 (coding) | 0.0010 | ||||||
| Hd6 | 2280 | 627 | 14 (5′), 0 (coding) | |||||||||
| Dst | 1031 | 927 | 3 (5′), 3 (coding) f | 0.0029 | 0.0032 | |||||||
| CHI | 1400 | 762 | 3 (5′), 1 (coding) | 0 (5′), 1 (coding) | 0.0013 | 5 (5′), 0 (coding) f | 0.0036 | |||||
| 4 | GIF1 | 1238 | 1794 | 2 (5′), 3 (1 coding) | 0.0006 | |||||||
| Myb4 | 1008 | 789 | 3 (5′), 3 (coding) | 0.0030 | 0.0038 | |||||||
| An-2 | 1000 | 753 | 4 (5′), 0 (coding) | 0.0040 | ||||||||
| unknown | 1010 | 1695 | 0 (5′), 1 (coding) | 0.0006 | 1 (5′), 2 (coding) f | 0.0010 | 0.0012 | |||||
| AGO2 | 1094 | 3123 | 2 (5′), 1 (coding) | 0.0003 | 4 (5′), 6 (coding) f | 0.0037 | 0.0019 | |||||
| IPK1 a | 1000 | 1338 | 0 (5′), 1 (coding) | 0.0007 | 2 (5′), 0 (coding) f | 0.0020 | ||||||
| F3H | 1002 | 1134 | 3 (5′), 1 (coding) | 0.0030 | 0.0009 | 3 (5′), 4 (coding) f | 0.0030 | 0.0035 | ||||
| SH4 | 1006 | 1173 | 5 (5′), 1 (5′), 1 (coding) | 0.0050 | 0.0009 | 1 (5′), 0 (coding) | 0.0010 | |||||
| 5 | GS5 | 1010 | 1458 | 1 (5′), 1 (coding) | 1 (5′), 2 (coding) f | 0.0010 | 0.0014 | |||||
| ACS3 | 1000 | 1314 | 1 (5′), 0 (coding) f | 0.0010 | ||||||||
| SH5 | 1466 | 1743 | 3 (5′), 3 (coding) f | 0.0020 | 0.0017 | |||||||
| T6P (TPS1) | 1018 | 2595 | 5 (5′), 4 (coding) f | 0.0049 | 0.0015 | |||||||
| 6 | EPSPS | 1007 | 1548 | 3 (5′), 0 (coding) | 0 (5′), 1 (coding) | 0.0006 | ||||||
| Hd3a | 1023 | 540 | 4 (5′), 0 (coding) | 0 (5′), 2 (coding) | 0.0037 | |||||||
| C1 | 1002 | 819 | 2 (5′), 0 (coding) | 0.0020 | ||||||||
| TCP19 | 1116 | 1209 | 1 (5′), 1 (5′), 0 (coding) | 0.0009 | 1 (5′), 0 (coding) | 0.0009 | ||||||
| Hd1 | 1375 | 1351 | 1 (5′), 2 (1 coding) | 0.0007 | ||||||||
| 7 | PROG1 | 1000 | 504 | 1 (5′), 1 (5′), 1 (coding) | 0.0010 | 0.0020 | ||||||
| Rc | 594 | 2013 | 1 (5′), 2 (5′), 3 (coding) | 0.0017 | 1 (5′), 0 (coding) | 0.0017 | ||||||
| SPL13 a | 1014 | 651 | 1 (5′), 0 (coding) e | 0.0010 | ||||||||
| WG7 a | 1064 | 4803 | 8 (5′), 4 (coding) | |||||||||
| 8 | RAE2 | 1003 | 593 | 3 (5′), 1 (coding) | 0.0030 | 0.0017 | ||||||
| SPL16 | 1017 | 1368 | 3 (5′), 0 (coding) | 1 (5′), 1 (coding) f | 0.0010 | 0.0007 | ||||||
| 9 | unknown | 1075 | 360 | 4 (5′), 2 (coding) f | 0.0037 | 0.0056 | ||||||
| DEP1 | 1000 | 1281 | 0 (5′), 2 (coding) | 0.0016 | 4 (5′), 0 (coding) f | 0.0040 | ||||||
| PGI | 1000 | 1878 | 2 (5′), 0 (coding) | 0.0020 | ||||||||
| PRR95 | 1034 | 1872 | 1 (5′), 1 (coding) | 0.0010 | 1 (5′), 1 (coding) f | 0.0010 | 0.0005 | |||||
| DHQS | 1013 | 1332 | 1 (5′), 0 (coding) f | 0.0010 | ||||||||
| 10 | DAHPS2 | 1007 | 1509 | 5 (5′), 1 (coding) | 0.0050 | 0.0007 | ||||||
| MYC2 | 1015 | 2261 | 1 (5′), 0 (coding) | 0.0010 | 2 (5′), 0 (coding) f | 0.0020 | ||||||
| 11 | PK1 | 1016 | 1584 | 2 (5′), 0 (coding) | 6 (5′), 0 (coding) | 0.0059 | ||||||
| unknown | 1004 | 1782 | 8 (5′), 0 (coding) | 0.0080 | ||||||||
| CHS | 1254 | 1197 | 1 (5′), 2 (coding) | 0.0008 | 0.0017 | 1 (5′), 0 (coding) | 0.0008 | |||||
| 12 | Pi-ta a | 1277 | 2787 | 1 (5′), 0 (coding) | 0.0008 | |||||||
| unknown | 1013 | 1348 | 2 (5′), 3 (coding) | 0.0022 | ||||||||
| Subgroups | Gene 1 a | Gene 2 | Gene 3 | Gene 4 | Gene 5 | Gene 6 | Gene 7 | Gene 8 |
|---|---|---|---|---|---|---|---|---|
| aus-aromatic | GIF1 5′(2) | RAE2 cds (3) b | FUWA 5′(5) | TAC3 5′(1) c | SPL13-5′ (1) c | |||
| GIF1 cds (1) | FUWA cds (1) | |||||||
| tropical-aromatic | CKX2 5′(1) | DFR 5′(12) | Hd3a 5′(3) | Hd1 5′(1) | Rc 5′(1) | SK2 cds (1) | PRR95 cds (1) | Os09g26890 5′(1) |
| CKX2 cds (1) | ||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y. Gene Genealogy-Based Mutation Analysis Reveals Emergence of Aus, Tropical japonica, and Aromatic of Oryza sativa during the Later Stage of Rice Domestication. Genes 2023, 14, 1412. https://doi.org/10.3390/genes14071412
Lu Y. Gene Genealogy-Based Mutation Analysis Reveals Emergence of Aus, Tropical japonica, and Aromatic of Oryza sativa during the Later Stage of Rice Domestication. Genes. 2023; 14(7):1412. https://doi.org/10.3390/genes14071412
Chicago/Turabian StyleLu, Yingqing. 2023. "Gene Genealogy-Based Mutation Analysis Reveals Emergence of Aus, Tropical japonica, and Aromatic of Oryza sativa during the Later Stage of Rice Domestication" Genes 14, no. 7: 1412. https://doi.org/10.3390/genes14071412
APA StyleLu, Y. (2023). Gene Genealogy-Based Mutation Analysis Reveals Emergence of Aus, Tropical japonica, and Aromatic of Oryza sativa during the Later Stage of Rice Domestication. Genes, 14(7), 1412. https://doi.org/10.3390/genes14071412