
Therapeutic Targeting of RNA Splicing in Cancer
Abstract
1. Introduction
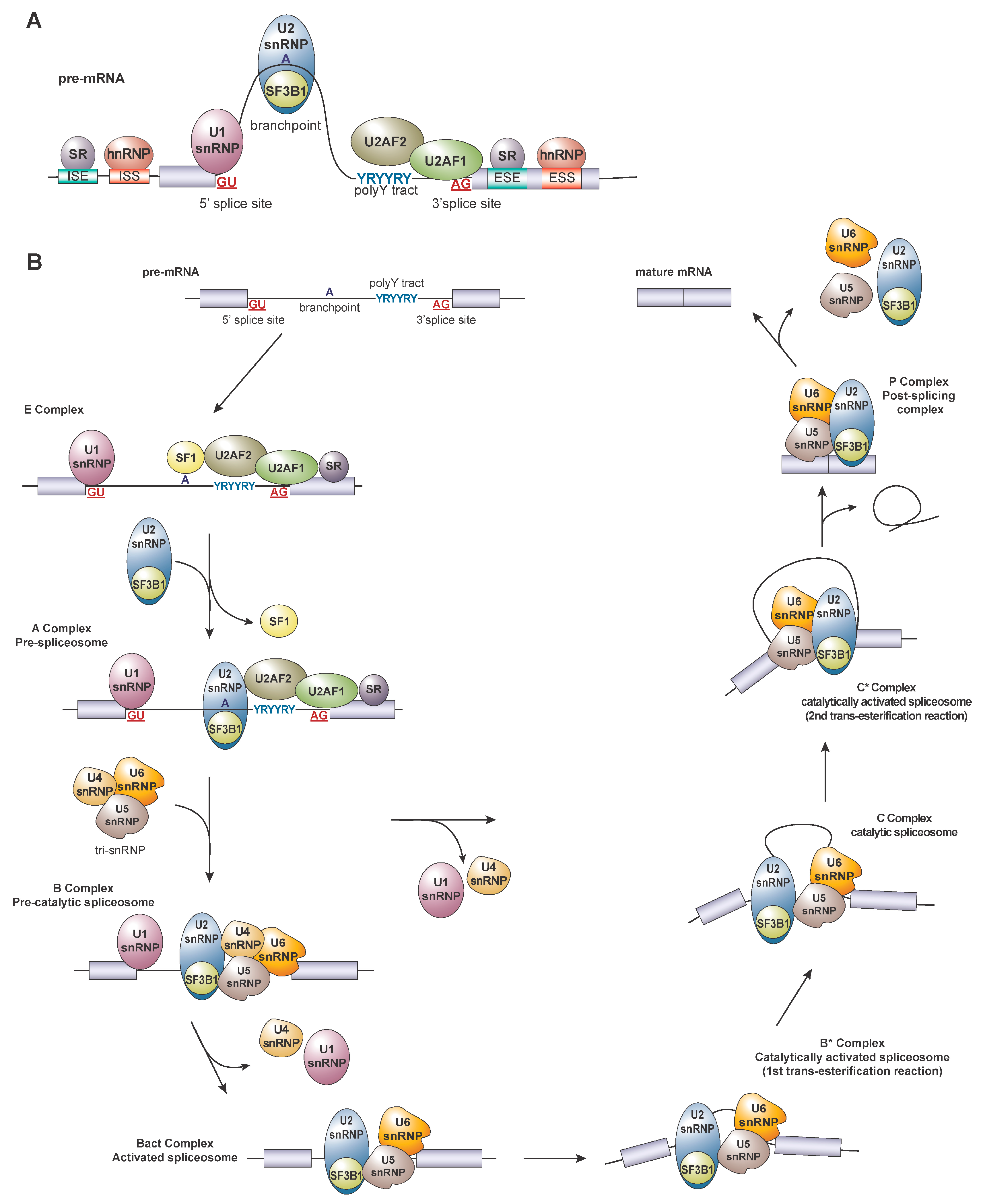
2. Regulation of Splicing
3. Altered Splicing Factor Expression in Cancer
4. Recurrent Mutations in Splicing Factors in Cancer
4.1. SF3B1 Mutations
4.2. U2AF1
4.3. SRSF2
4.4. ZRSR2
4.5. Other Spliceosome Components
4.6. Splicing in Metastasis and Treatment Resistance
5. Therapeutic Targeting of RNA Splicing in Cancer
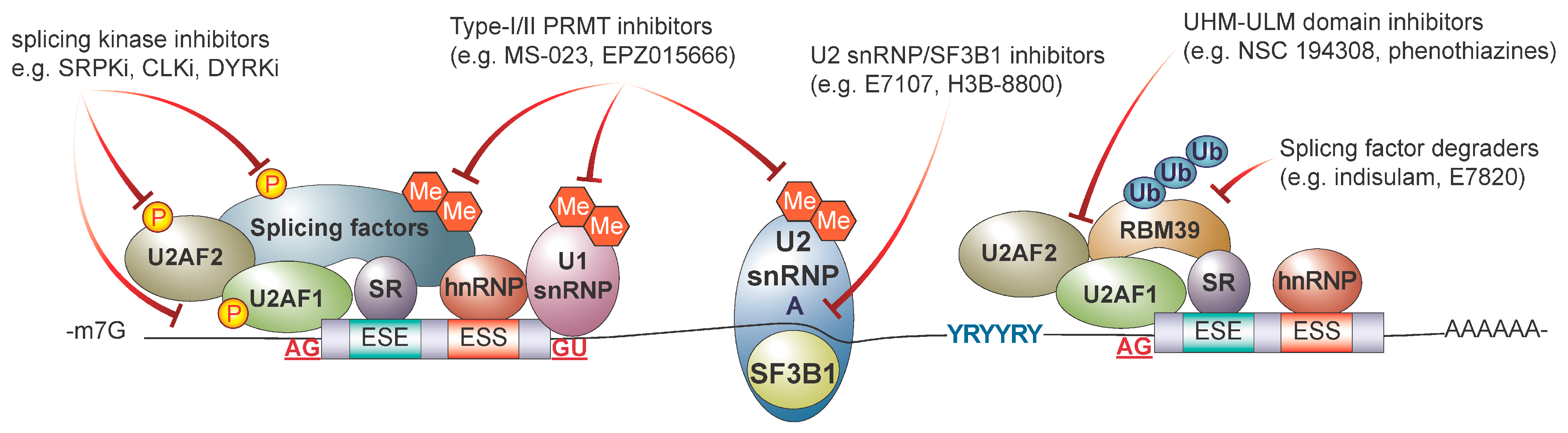
5.1. Targeting the Core Spliceosome with Small Molecule Inhibitors
5.2. Targeting Splicing Regulatory Proteins
5.3. Targeting RNA Binding Proteins
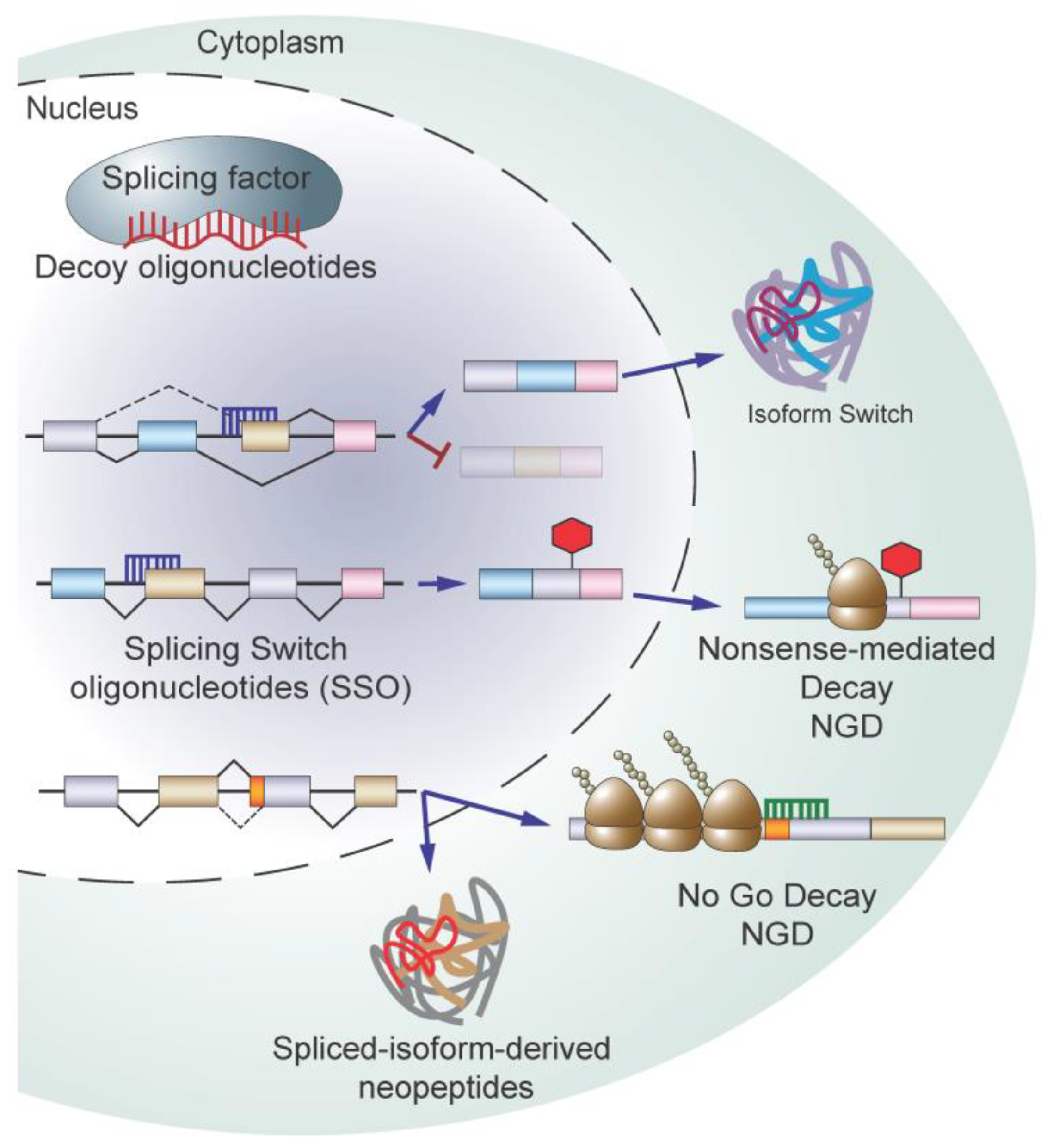
5.4. Targeting Splicing Using Oligonucleotide-Based Therapy
5.5. Immunotherapeutic Approaches Targeting RNA Splicing
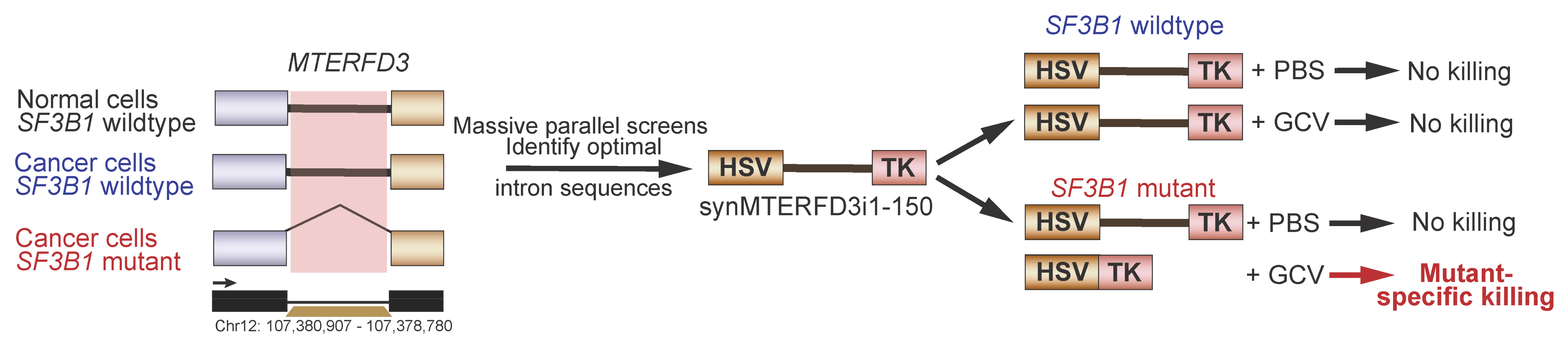
5.6. Synthetic Introns for Mutation-Specific Gene Expression
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wilkinson, M.E.; Charenton, C.; Nagai, K. RNA Splicing by the Spliceosome. Annu. Rev. Biochem. 2020, 89, 359–388. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.D.; Ares, M., Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Lee, D.; Lee, J.; Park, D.; Kim, Y.J.; Park, W.Y.; Hong, D.; Park, P.J.; Lee, E. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat. Genet. 2015, 47, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Miñana, B.; Valcárcel, J.; Gabaldón, T.; Lehner, B. Synonymous mutations frequently act as driver mutations in human cancers. Cell 2014, 156, 1324–1335. [Google Scholar] [CrossRef]
- Anczukow, O.; Rosenberg, A.Z.; Akerman, M.; Das, S.; Zhan, L.; Karni, R.; Muthuswamy, S.K.; Krainer, A.R. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012, 19, 220–228. [Google Scholar] [CrossRef]
- Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef]
- Ben-Hur, V.; Denichenko, P.; Siegfried, Z.; Maimon, A.; Krainer, A.; Davidson, B.; Karni, R. S6K1 alternative splicing modulates its oncogenic activity and regulates mTORC1. Cell Rep. 2013, 3, 103–115. [Google Scholar] [CrossRef]
- Du, J.X.; Luo, Y.H.; Zhang, S.J.; Wang, B.; Chen, C.; Zhu, G.Q.; Zhu, P.; Cai, C.Z.; Wan, J.L.; Cai, J.L.; et al. Splicing factor SRSF1 promotes breast cancer progression via oncogenic splice switching of PTPMT1. J. Exp. Clin. Cancer Res. 2021, 40, 171. [Google Scholar] [CrossRef]
- Bradley, R.K.; Anczukow, O. RNA splicing dysregulation and the hallmarks of cancer. Nat. Rev. Cancer 2023, 23, 135–155. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef]
- Chen, S.; Benbarche, S.; Abdel-Wahab, O. Splicing factor mutations in hematologic malignancies. Blood 2021, 138, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef]
- Bertram, K.; Agafonov, D.E.; Dybkov, O.; Haselbach, D.; Leelaram, M.N.; Will, C.L.; Urlaub, H.; Kastner, B.; Luhrmann, R.; Stark, H. Cryo-EM Structure of a Pre-catalytic Human Spliceosome Primed for Activation. Cell 2017, 170, 701–713.e711. [Google Scholar] [CrossRef] [PubMed]
- Kielkopf, C.L. Insights from structures of cancer-relevant pre-mRNA splicing factors. Curr. Opin. Genet. Dev. 2018, 48, 57–66. [Google Scholar] [CrossRef]
- Plaschka, C.; Lin, P.C.; Nagai, K. Structure of a pre-catalytic spliceosome. Nature 2017, 546, 617–621. [Google Scholar] [CrossRef]
- Yan, C.; Hang, J.; Wan, R.; Huang, M.; Wong, C.C.; Shi, Y. Structure of a yeast spliceosome at 3.6-angstrom resolution. Science 2015, 349, 1182–1191. [Google Scholar] [CrossRef] [PubMed]
- Clough, C.A.; Pangallo, J.; Sarchi, M.; Ilagan, J.O.; North, K.; Bergantinos, R.; Stolla, M.C.; Naru, J.; Nugent, P.; Kim, E.; et al. Coordinated missplicing of TMEM14C and ABCB7 causes ring sideroblast formation in SF3B1-mutant myelodysplastic syndrome. Blood 2022, 139, 2038–2049. [Google Scholar] [CrossRef]
- Inoue, D.; Chew, G.L.; Liu, B.; Michel, B.C.; Pangallo, J.; D’Avino, A.R.; Hitchman, T.; North, K.; Lee, S.C.; Bitner, L.; et al. Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature 2019, 574, 432–436. [Google Scholar] [CrossRef]
- Liu, Z.; Yoshimi, A.; Wang, J.; Cho, H.; Chun-Wei Lee, S.; Ki, M.; Bitner, L.; Chu, T.; Shah, H.; Liu, B.; et al. Mutations in the RNA Splicing Factor SF3B1 Promote Tumorigenesis through MYC Stabilization. Cancer Discov. 2020, 10, 806–821. [Google Scholar] [CrossRef]
- Wu, S.; Romfo, C.M.; Nilsen, T.W.; Green, M.R. Functional recognition of the 3′ splice site AG by the splicing factor U2AF35. Nature 1999, 402, 832–835. [Google Scholar] [CrossRef] [PubMed]
- Ilagan, J.O.; Ramakrishnan, A.; Hayes, B.; Murphy, M.E.; Zebari, A.S.; Bradley, P.; Bradley, R.K. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015, 25, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Yip, B.H.; Steeples, V.; Repapi, E.; Armstrong, R.N.; Llorian, M.; Roy, S.; Shaw, J.; Dolatshad, H.; Taylor, S.; Verma, A.; et al. The U2AF1S34F mutation induces lineage-specific splicing alterations in myelodysplastic syndromes. J. Clin. Investig. 2017, 127, 2206–2221. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef]
- Lee, S.C.; North, K.; Kim, E.; Jang, E.; Obeng, E.; Lu, S.X.; Liu, B.; Inoue, D.; Yoshimi, A.; Ki, M.; et al. Synthetic Lethal and Convergent Biological Effects of Cancer-Associated Spliceosomal Gene Mutations. Cancer Cell 2018, 34, 225–241.e228. [Google Scholar] [CrossRef]
- Madan, V.; Kanojia, D.; Li, J.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A.; Sanada, M.; Grossmann, V.; Sundaresan, J.; Shiraishi, Y.; et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat. Commun. 2015, 6, 6042. [Google Scholar] [CrossRef]
- Inoue, D.; Polaski, J.T.; Taylor, J.; Castel, P.; Chen, S.; Kobayashi, S.; Hogg, S.J.; Hayashi, Y.; Pineda, J.M.B.; El Marabti, E.; et al. Minor intron retention drives clonal hematopoietic disorders and diverse cancer predisposition. Nat. Genet. 2021, 53, 707–718. [Google Scholar] [CrossRef]
- Polprasert, C.; Schulze, I.; Sekeres, M.A.; Makishima, H.; Przychodzen, B.; Hosono, N.; Singh, J.; Padgett, R.A.; Gu, X.; Phillips, J.G.; et al. Inherited and Somatic Defects in DDX41 in Myeloid Neoplasms. Cancer Cell 2015, 27, 658–670. [Google Scholar] [CrossRef]
- Daniels, N.J.; Hershberger, C.E.; Gu, X.; Schueger, C.; DiPasquale, W.M.; Brick, J.; Saunthararajah, Y.; Maciejewski, J.P.; Padgett, R.A. Functional analyses of human LUC7-like proteins involved in splicing regulation and myeloid neoplasms. Cell Rep. 2021, 35, 108989. [Google Scholar] [CrossRef]
- Makishima, H.; Visconte, V.; Sakaguchi, H.; Jankowska, A.M.; Abu Kar, S.; Jerez, A.; Przychodzen, B.; Bupathi, M.; Guinta, K.; Afable, M.G.; et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 2012, 119, 3203–3210. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Tomsic, J.; He, H.; Akagi, K.; Liyanarachchi, S.; Pan, Q.; Bertani, B.; Nagy, R.; Symer, D.E.; Blencowe, B.J.; de la Chapelle, A. A germline mutation in SRRM2, a splicing factor gene, is implicated in papillary thyroid carcinoma predisposition. Sci. Rep. 2015, 5, 10566. [Google Scholar] [CrossRef]
- Lu, Z.X.; Huang, Q.; Park, J.W.; Shen, S.; Lin, L.; Tokheim, C.J.; Henry, M.D.; Xing, Y. Transcriptome-wide landscape of pre-mRNA alternative splicing associated with metastatic colonization. Mol. Cancer Res. 2015, 13, 305–318. [Google Scholar] [CrossRef]
- Bebee, T.W.; Park, J.W.; Sheridan, K.I.; Warzecha, C.C.; Cieply, B.W.; Rohacek, A.M.; Xing, Y.; Carstens, R.P. The splicing regulators Esrp1 and Esrp2 direct an epithelial splicing program essential for mammalian development. Elife 2015, 4, e08954. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Du, C.; Kuang, S.; Zhou, X.; Zhang, J.; Ao, X. Underlying mechanisms of epithelial splicing regulatory proteins in cancer progression. J. Mol. Med. 2022, 100, 1539–1556. [Google Scholar] [CrossRef]
- Venables, J.P.; Brosseau, J.P.; Gadea, G.; Klinck, R.; Prinos, P.; Beaulieu, J.F.; Lapointe, E.; Durand, M.; Thibault, P.; Tremblay, K.; et al. RBFOX2 is an important regulator of mesenchymal tissue-specific splicing in both normal and cancer tissues. Mol. Cell Biol. 2013, 33, 396–405. [Google Scholar] [CrossRef]
- Jbara, A.; Lin, K.T.; Stossel, C.; Siegfried, Z.; Shqerat, H.; Amar-Schwartz, A.; Elyada, E.; Mogilevsky, M.; Raitses-Gurevich, M.; Johnson, J.L.; et al. RBFOX2 modulates a metastatic signature of alternative splicing in pancreatic cancer. Nature 2023, 617, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Ghigna, C.; Giordano, S.; Shen, H.; Benvenuto, F.; Castiglioni, F.; Comoglio, P.M.; Green, M.R.; Riva, S.; Biamonti, G. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol. Cell 2005, 20, 881–890. [Google Scholar] [CrossRef]
- Lin, J.C.; Lee, Y.C.; Tan, T.H.; Liang, Y.C.; Chuang, H.C.; Fann, Y.C.; Johnson, K.R.; Lin, Y.J. RBM4-SRSF3-MAP4K4 splicing cascade modulates the metastatic signature of colorectal cancer cell. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, I.M.; Cheng, A.W.; Flytzanis, N.C.; Balsamo, M.; Condeelis, J.S.; Oktay, M.H.; Burge, C.B.; Gertler, F.B. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet. 2011, 7, e1002218. [Google Scholar] [CrossRef] [PubMed]
- Gautrey, H.; Jackson, C.; Dittrich, A.L.; Browell, D.; Lennard, T.; Tyson-Capper, A. SRSF3 and hnRNP H1 regulate a splicing hotspot of HER2 in breast cancer cells. RNA Biol. 2015, 12, 1139–1151. [Google Scholar] [CrossRef]
- Jiang, P.; Li, Z.; Tian, F.; Li, X.; Yang, J. Fyn/heterogeneous nuclear ribonucleoprotein E1 signaling regulates pancreatic cancer metastasis by affecting the alternative splicing of integrin beta1. Int. J. Oncol. 2017, 51, 169–183. [Google Scholar] [CrossRef]
- Loh, T.J.; Moon, H.; Cho, S.; Jang, H.; Liu, Y.C.; Tai, H.; Jung, D.W.; Williams, D.R.; Kim, H.R.; Shin, M.G.; et al. CD44 alternative splicing and hnRNP A1 expression are associated with the metastasis of breast cancer. Oncol. Rep. 2015, 34, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.P.; Hillmer, A.M.; Chuah, C.T.; Juan, W.C.; Ko, T.K.; Teo, A.S.; Ariyaratne, P.N.; Takahashi, N.; Sawada, K.; Fei, Y.; et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat. Med. 2012, 18, 521–528. [Google Scholar] [CrossRef]
- Stark, M.; Bram, E.E.; Akerman, M.; Mandel-Gutfreund, Y.; Assaraf, Y.G. Heterogeneous nuclear ribonucleoprotein H1/H2-dependent unsplicing of thymidine phosphorylase results in anticancer drug resistance. J. Biol. Chem. 2011, 286, 3741–3754. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Lopez, M.; Schulz, L.; Enculescu, M.; Paret, C.; Spiekermann, B.; Quesnel-Vallieres, M.; Torres-Diz, M.; Unic, S.; Busch, A.; Orekhova, A.; et al. High-throughput mutagenesis identifies mutations and RNA-binding proteins controlling CD19 splicing and CART-19 therapy resistance. Nat. Commun. 2022, 13, 5570. [Google Scholar] [CrossRef]
- Fischer, J.; Paret, C.; El Malki, K.; Alt, F.; Wingerter, A.; Neu, M.A.; Kron, B.; Russo, A.; Lehmann, N.; Roth, L.; et al. CD19 Isoforms Enabling Resistance to CART-19 Immunotherapy Are Expressed in B-ALL Patients at Initial Diagnosis. J. Immunother. 2017, 40, 187–195. [Google Scholar] [CrossRef]
- Nadiminty, N.; Tummala, R.; Liu, C.; Lou, W.; Evans, C.P.; Gao, A.C. NF-kappaB2/p52:c-Myc:hnRNPA1 Pathway Regulates Expression of Androgen Receptor Splice Variants and Enzalutamide Sensitivity in Prostate Cancer. Mol. Cancer Ther. 2015, 14, 1884–1895. [Google Scholar] [CrossRef]
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; van der Gulden, H.; van der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-Delta11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 2016, 76, 2778–2790. [Google Scholar] [CrossRef] [PubMed]
- Ozden, O.; Bishehsari, F.; Bauer, J.; Park, S.H.; Jana, A.; Baik, S.H.; Sporn, J.C.; Staudacher, J.J.; Yazici, C.; Krett, N.; et al. Expression of an Oncogenic BARD1 Splice Variant Impairs Homologous Recombination and Predicts Response to PARP-1 Inhibitor Therapy in Colon Cancer. Sci. Rep. 2016, 6, 26273. [Google Scholar] [CrossRef] [PubMed]
- Lai, F.; Jiang, C.C.; Farrelly, M.L.; Zhang, X.D.; Hersey, P. Evidence for upregulation of Bim and the splicing factor SRp55 in melanoma cells from patients treated with selective BRAF inhibitors. Melanoma Res. 2012, 22, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef]
- Castiglioni, F.; Tagliabue, E.; Campiglio, M.; Pupa, S.M.; Balsari, A.; Menard, S. Role of exon-16-deleted HER2 in breast carcinomas. Endocr. Relat. Cancer 2006, 13, 221–232. [Google Scholar] [CrossRef]
- Ang, Z.; Hayer, K.E.; Diz, M.T.; Schmidt, C.; Zheng, S.; Xu, F.; Falkenstein, C.D.; Loftus, J.P.; Yang, S.Y.; Asnani, M.; et al. Alternative splicing of its 5’ untranslated region controls CD20 mRNA translation and enables resistance to CD20-directed immunotherapies. bioRxiv 2023. [Google Scholar] [CrossRef]
- Tripathi, V.; Shin, J.H.; Stuelten, C.H.; Zhang, Y.E. TGF-beta-induced alternative splicing of TAK1 promotes EMT and drug resistance. Oncogene 2019, 38, 3185–3200. [Google Scholar] [CrossRef]
- Calabretta, S.; Bielli, P.; Passacantilli, I.; Pilozzi, E.; Fendrich, V.; Capurso, G.; Fave, G.D.; Sette, C. Modulation of PKM alternative splicing by PTBP1 promotes gemcitabine resistance in pancreatic cancer cells. Oncogene 2016, 35, 2031–2039. [Google Scholar] [CrossRef]
- Nakajima, H.; Sato, B.; Fujita, T.; Takase, S.; Terano, H.; Okuhara, M. New antitumor substances, FR901463, FR901464 and FR901465. I. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities. J. Antibiot. 1996, 49, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Kaida, D.; Motoyoshi, H.; Tashiro, E.; Nojima, T.; Hagiwara, M.; Ishigami, K.; Watanabe, H.; Kitahara, T.; Yoshida, T.; Nakajima, H.; et al. Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat. Chem. Biol. 2007, 3, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Corrionero, A.; Minana, B.; Valcarcel, J. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev. 2011, 25, 445–459. [Google Scholar] [CrossRef]
- Abelson, S.; Collord, G.; Ng, S.W.K.; Weissbrod, O.; Mendelson Cohen, N.; Niemeyer, E.; Barda, N.; Zuzarte, P.C.; Heisler, L.; Sundaravadanam, Y.; et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature 2018, 559, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Sakai, Y.; Tsujita, T.; Akiyama, T.; Yoshida, T.; Mizukami, T.; Akinaga, S.; Horinouchi, S.; Yoshida, M.; Yoshida, T. GEX1 compounds, novel antitumor antibiotics related to herboxidiene, produced by Streptomyces sp. II. The effects on cell cycle progression and gene expression. J. Antibiot. 2002, 55, 863–872. [Google Scholar] [CrossRef]
- Sellin, M.; Mack, R.; Rhodes, M.C.; Zhang, L.; Berg, S.; Joshi, K.; Liu, S.; Wei, W.; S, J.P.; Larsen, P.; et al. Molecular mechanisms by which splice modulator GEX1A inhibits leukaemia development and progression. Br. J. Cancer 2022, 127, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Ozawa, Y.; Shimizu, H.; Niijima, J.; Kanada, R.; Nagai, M.; Kotake, Y.; Yoshida, M.; Tsuchida, T.; Mizui, Y.; et al. E7107, a new 7-urethane derivative of pladienolide D, displays curative effect against several human tumor xenografts. Cancer Res. 2004, 45, 691. [Google Scholar]
- Mizui, Y.; Sakai, T.; Iwata, M.; Uenaka, T.; Okamoto, K.; Shimizu, H.; Yamori, T.; Yoshimatsu, K.; Asada, M. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities. J. Antibiot. 2004, 57, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Sagane, K.; Owa, T.; Mimori-Kiyosue, Y.; Shimizu, H.; Uesugi, M.; Ishihama, Y.; Iwata, M.; Mizui, Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 2007, 3, 570–575. [Google Scholar] [CrossRef]
- Albert, B.J.; Sivaramakrishnan, A.; Naka, T.; Czaicki, N.L.; Koide, K. Total syntheses, fragmentation studies, and antitumor/antiproliferative activities of FR901464 and its low picomolar analogue. J. Am. Chem. Soc. 2007, 129, 2648–2659. [Google Scholar] [CrossRef]
- Eskens, F.A.; Ramos, F.J.; Burger, H.; O’Brien, J.P.; Piera, A.; de Jonge, M.J.; Mizui, Y.; Wiemer, E.A.; Carreras, M.J.; Baselga, J.; et al. Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin. Cancer Res. 2013, 19, 6296–6304. [Google Scholar] [CrossRef]
- Hong, D.S.; Kurzrock, R.; Naing, A.; Wheler, J.J.; Falchook, G.S.; Schiffman, J.S.; Faulkner, N.; Pilat, M.J.; O’Brien, J.; LoRusso, P. A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Investig. New Drugs 2014, 32, 436–444. [Google Scholar] [CrossRef]
- Kanada, R.M.; Itoh, D.; Nagai, M.; Niijima, J.; Asai, N.; Mizui, Y.; Abe, S.; Kotake, Y. Total synthesis of the potent antitumor macrolides pladienolide B and D. Angew Chem. Int. Ed. Engl. 2007, 46, 4350–4355. [Google Scholar] [CrossRef]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Dvinge, H.; Kim, E.; Cho, H.; Micol, J.B.; Chung, Y.R.; Durham, B.H.; Yoshimi, A.; Kim, Y.J.; Thomas, M.; et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat. Med. 2016, 22, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G.; et al. Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 2016, 30, 404–417. [Google Scholar] [CrossRef] [PubMed]
- Shirai, C.L.; White, B.S.; Tripathi, M.; Tapia, R.; Ley, J.N.; Ndonwi, M.; Kim, S.; Shao, J.; Carver, A.; Saez, B.; et al. Mutant U2AF1-expressing cells are sensitive to pharmacological modulation of the spliceosome. Nat. Commun. 2017, 8, 14060. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L.; et al. Phase I First-in-Human Dose Escalation Study of the oral SF3B1 modulator H3B-8800 in myeloid neoplasms. Leukemia 2021, 35, 3542–3550. [Google Scholar] [CrossRef]
- Chatrikhi, R.; Feeney, C.F.; Pulvino, M.J.; Alachouzos, G.; MacRae, A.J.; Falls, Z.; Rai, S.; Brennessel, W.W.; Jenkins, J.L.; Walter, M.J.; et al. A synthetic small molecule stalls pre-mRNA splicing by promoting an early-stage U2AF2-RNA complex. Cell Chem. Biol. 2021, 28, 1145–1157.e1146. [Google Scholar] [CrossRef]
- Jagtap, P.K.A.; Kubelka, T.; Soni, K.; Will, C.L.; Garg, D.; Sippel, C.; Kapp, T.G.; Potukuchi, H.K.; Schorpp, K.; Hadian, K.; et al. Identification of phenothiazine derivatives as UHM-binding inhibitors of early spliceosome assembly. Nat. Commun. 2020, 11, 5621. [Google Scholar] [CrossRef]
- Giannakouros, T.; Nikolakaki, E.; Mylonis, I.; Georgatsou, E. Serine-arginine protein kinases: A small protein kinase family with a large cellular presence. FEBS J. 2011, 278, 570–586. [Google Scholar] [CrossRef]
- Siqueira, R.P.; Barbosa Ede, A.; Poleto, M.D.; Righetto, G.L.; Seraphim, T.V.; Salgado, R.L.; Ferreira, J.G.; Barros, M.V.; de Oliveira, L.L.; Laranjeira, A.B.; et al. Potential Antileukemia Effect and Structural Analyses of SRPK Inhibition by N-(2-(Piperidin-1-yl)-5-(Trifluoromethyl)Phenyl)Isonicotinamide (SRPIN340). PLoS One 2015, 10, e0134882. [Google Scholar] [CrossRef]
- Tzelepis, K.; Koike-Yusa, H.; De Braekeleer, E.; Li, Y.; Metzakopian, E.; Dovey, O.M.; Mupo, A.; Grinkevich, V.; Li, M.; Mazan, M.; et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016, 17, 1193–1205. [Google Scholar] [CrossRef]
- Iwai, K.; Yaguchi, M.; Nishimura, K.; Yamamoto, Y.; Tamura, T.; Nakata, D.; Dairiki, R.; Kawakita, Y.; Mizojiri, R.; Ito, Y.; et al. Anti-tumor efficacy of a novel CLK inhibitor via targeting RNA splicing and MYC-dependent vulnerability. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Yoda, A.; Morishita, D.; Ochi, Y.; Mizutani, A.; Takeda, J.; Tozaki, H.; Satoh, Y.; Nannya, Y.; Makishima, H.; Miyake, H.; et al. CTX-712, a Novel Clk Inhibitor Targeting Myeloid Neoplasms with SRSF2 Mutation. Blood 2021, 138. [Google Scholar] [CrossRef]
- Shimizu, T.; Yonemori, K.; Koyama, T.; Katsuya, Y.; Sato, J.; Fukuhara, N.; Yokoyama, H.; Iida, H.; Ando, K.; Fukuhara, S.; et al. A first-in-human phase I study of CTX-712 in patients with advanced, relapsed or refractory malignant tumors. J. Clin. Oncol. 2022, 40, 3080. [Google Scholar] [CrossRef]
- Song, M.; Pang, L.; Zhang, M.; Qu, Y.; Laster, K.V.; Dong, Z. Cdc2-like kinases: Structure, biological function, and therapeutic targets for diseases. Signal Transduct Target Ther. 2023, 8, 148. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Zhao, J.; Pearson, Z.J.; Boskovic, Z.V.; Wang, J. RNA-Targeting Splicing Modifiers: Drug Development and Screening Assays. Molecules 2021, 26, 2263. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhang, T.; Zhou, C.; Chohan, M.O.; Gu, X.; Wegiel, J.; Zhou, J.; Hwang, Y.W.; Iqbal, K.; Grundke-Iqbal, I.; et al. Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of tau in Down syndrome. J. Biol. Chem. 2008, 283, 28660–28669. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.J.; Bhansali, R.; Diebold, L.; Cook, D.E.; Stolzenburg, L.; Casagrande, A.S.; Besson, T.; Leblond, B.; Desire, L.; Malinge, S.; et al. DYRK1A controls the transition from proliferation to quiescence during lymphoid development by destabilizing Cyclin D3. J. Exp. Med. 2015, 212, 953–970. [Google Scholar] [CrossRef]
- Rammohan, M.; Harris, E.; Bhansali, R.S.; Zhao, E.; Li, L.S.; Crispino, J.D. The chromosome 21 kinase DYRK1A: Emerging roles in cancer biology and potential as a therapeutic target. Oncogene 2022, 41, 2003–2011. [Google Scholar] [CrossRef]
- Bossard, C.; McMillan, E.A.; Creger, E.; Eastman, B.; Mak, C.-C.; Beaupre, D.M.; White, M.A. The Pan-Clk/Dyrk Inhibitor Cirtuvivint (SM08502) Exposes Mechanistic Underpinnings of Alternative Splicing As a Therapeutic Vulnerability in Heme Malignancies. Blood 2021, 138, 2950. [Google Scholar] [CrossRef]
- Jarvis, M.; Mittapalli, G.; Creger, E.; Nora, C.; Ibanez, M.; Bhat, D.; Hofilena, B.; Stewart, J.; McMillan, E.A.; Falahatpisheh, H.; et al. Next-generation DYRK1A inhibitors as a new therapeutic approach for the treatment of hematological malignancies. Cancer Res. 2023, 83, 5019. [Google Scholar] [CrossRef]
- Wang, E.; Pineda, J.M.B.; Kim, W.J.; Chen, S.; Bourcier, J.; Stahl, M.; Hogg, S.J.; Bewersdorf, J.P.; Han, C.; Singer, M.E.; et al. Modulation of RNA splicing enhances response to BCL2 inhibition in leukemia. Cancer Cell 2023, 41, 164–180.e168. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.Q.; Li, Y.; Yang, C.; Li, J.; Tan, S.H.; Chin, D.W.L.; Nakamura-Ishizu, A.; Yang, H.; Suda, T. PRMT5 Modulates Splicing for Genome Integrity and Preserves Proteostasis of Hematopoietic Stem Cells. Cell Rep. 2019, 26, 2316–2328.e2316. [Google Scholar] [CrossRef]
- Koh, C.M.; Bezzi, M.; Low, D.H.; Ang, W.X.; Teo, S.X.; Gay, F.P.; Al-Haddawi, M.; Tan, S.Y.; Osato, M.; Sabo, A.; et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 2015, 523, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Fong, J.Y.; Pignata, L.; Goy, P.A.; Kawabata, K.C.; Lee, S.C.; Koh, C.M.; Musiani, D.; Massignani, E.; Kotini, A.G.; Penson, A.; et al. Therapeutic Targeting of RNA Splicing Catalysis through Inhibition of Protein Arginine Methylation. Cancer Cell 2019, 36, 194–209.e199. [Google Scholar] [CrossRef]
- Ito, K.; Thodima, V.; Carter, J.; Bhagwat, N.; Sivakumar, M.; Grego, A.; Rager, J.; Terai, M.; Sato, T.; Abdel-Wahab, O.; et al. PRMT5 inhibition regulates alternative splicing and DNA damage repair pathways in SF3B1 R625G expressing uveal melanoma cells. Cancer Res. 2021, 81. [Google Scholar] [CrossRef]
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Nijhawan, D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356, eaal3755. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Minoshima, Y.; Sagane, K.; Sugi, N.H.; Mitsuhashi, K.O.; Yamamoto, N.; Kamiyama, H.; Takahashi, K.; Kotake, Y.; Uesugi, M.; et al. Selective degradation of splicing factor CAPERalpha by anticancer sulfonamides. Nat. Chem. Biol. 2017, 13, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, K.; Usuda, J.; Iwamoto, Y.; Fukumoto, H.; Nakamura, T.; Yoneda, T.; Narita, N.; Saijo, N.; Nishio, K. Mechanisms of action of the novel sulfonamide anticancer agent E7070 on cell cycle progression in human non-small cell lung cancer cells. Investig. New Drugs 2001, 19, 219–227. [Google Scholar] [CrossRef]
- Owa, T.; Yoshino, H.; Okauchi, T.; Yoshimatsu, K.; Ozawa, Y.; Sugi, N.H.; Nagasu, T.; Koyanagi, N.; Kitoh, K. Discovery of novel antitumor sulfonamides targeting G1 phase of the cell cycle. J. Med. Chem. 1999, 42, 3789–3799. [Google Scholar] [CrossRef]
- Ozawa, Y.; Sugi, N.H.; Nagasu, T.; Owa, T.; Watanabe, T.; Koyanagi, N.; Yoshino, H.; Kitoh, K.; Yoshimatsu, K. E7070, a novel sulphonamide agent with potent antitumour activity in vitro and in vivo. Eur. J. Cancer 2001, 37, 2275–2282. [Google Scholar] [CrossRef]
- Imai, H.; Chan, E.K.; Kiyosawa, K.; Fu, X.D.; Tan, E.M. Novel nuclear autoantigen with splicing factor motifs identified with antibody from hepatocellular carcinoma. J. Clin. Investig. 1993, 92, 2419–2426. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G.; et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Nijhuis, A.; Keun, H.C. RNA-binding motif protein 39 (RBM39): An emerging cancer target. Br. J. Pharmacol. 2022, 179, 2795–2812. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, W.; Zhang, N.; Chen, X.; Liu, W.; Zhang, L.; Liu, N. Systematic pan-cancer analysis identifies RBM39 as an immunological and prognostic biomarker. J. Cell Mol. Med. 2022, 26, 4859–4871. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Lu, S.X.; Pastore, A.; Chen, X.; Imig, J.; Chun-Wei Lee, S.; Hockemeyer, K.; Ghebrechristos, Y.E.; Yoshimi, A.; Inoue, D.; et al. Targeting an RNA-Binding Protein Network in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 369–384.e367. [Google Scholar] [CrossRef] [PubMed]
- Dowhan, D.H.; Hong, E.P.; Auboeuf, D.; Dennis, A.P.; Wilson, M.M.; Berget, S.M.; O’Malley, B.W. Steroid hormone receptor coactivation and alternative RNA splicing by U2AF65-related proteins CAPERalpha and CAPERbeta. Mol. Cell 2005, 17, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Loerch, S.; Maucuer, A.; Manceau, V.; Green, M.R.; Kielkopf, C.L. Cancer-relevant splicing factor CAPERalpha engages the essential splicing factor SF3b155 in a specific ternary complex. J. Biol. Chem. 2014, 289, 17325–17337. [Google Scholar] [CrossRef]
- Stepanyuk, G.A.; Serrano, P.; Peralta, E.; Farr, C.L.; Axelrod, H.L.; Geralt, M.; Das, D.; Chiu, H.J.; Jaroszewski, L.; Deacon, A.M.; et al. UHM-ULM interactions in the RBM39-U2AF65 splicing-factor complex. Acta Cryst. D Struct. Biol. 2016, 72, 497–511. [Google Scholar] [CrossRef]
- Xiong, H.; Veedu, R.N.; Diermeier, S.D. Recent Advances in Oligonucleotide Therapeutics in Oncology. Int. J. Mol. Sci. 2021, 22, 3295. [Google Scholar] [CrossRef]
- Dominski, Z.; Kole, R. Restoration of correct splicing in thalassemic pre-mRNA by antisense oligonucleotides. Proc. Natl. Acad. Sci. USA 1993, 90, 8673–8677. [Google Scholar] [CrossRef]
- Sergeeva, O.V.; Shcherbinina, E.Y.; Shomron, N.; Zatsepin, T.S. Modulation of RNA Splicing by Oligonucleotides: Mechanisms of Action and Therapeutic Implications. Nucleic Acid Ther. 2022, 32, 123–138. [Google Scholar] [CrossRef]
- Ma, W.K.; Voss, D.M.; Scharner, J.; Costa, A.S.H.; Lin, K.T.; Jeon, H.Y.; Wilkinson, J.E.; Jackson, M.; Rigo, F.; Bennett, C.F.; et al. ASO-Based PKM Splice-Switching Therapy Inhibits Hepatocellular Carcinoma Growth. Cancer Res. 2022, 82, 900–915. [Google Scholar] [CrossRef] [PubMed]
- Dewaele, M.; Tabaglio, T.; Willekens, K.; Bezzi, M.; Teo, S.X.; Low, D.H.; Koh, C.M.; Rambow, F.; Fiers, M.; Rogiers, A.; et al. Antisense oligonucleotide-mediated MDM4 exon 6 skipping impairs tumor growth. J. Clin. Investig. 2016, 126, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Khurshid, S.; Montes, M.; Comiskey, D.F., Jr.; Shane, B.; Matsa, E.; Jung, F.; Brown, C.; Bid, H.K.; Wang, R.; Houghton, P.J.; et al. Splice-switching of the insulin receptor pre-mRNA alleviates tumorigenic hallmarks in rhabdomyosarcoma. NPJ Precis. Oncol. 2022, 6, 1. [Google Scholar] [CrossRef]
- Li, L.; Hobson, L.; Perry, L.; Clark, B.; Heavey, S.; Haider, A.; Sridhar, A.; Shaw, G.; Kelly, J.; Freeman, A.; et al. Targeting the ERG oncogene with splice-switching oligonucleotides as a novel therapeutic strategy in prostate cancer. Br. J. Cancer 2020, 123, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, Q.; Han, L.; Tian, N.; Liang, Q.; Li, Y.; Zhao, X.; Du, C.; Tian, Y. Pro-apoptotic effects of splice-switching oligonucleotides targeting Bcl-x pre-mRNA in human glioma cell lines. Oncol. Rep. 2016, 35, 1013–1019. [Google Scholar] [CrossRef]
- Thng, D.K.H.; Toh, T.B.; Pigini, P.; Hooi, L.; Dan, Y.Y.; Chow, P.K.; Bonney, G.K.; Rashid, M.; Guccione, E.; Wee, D.K.B.; et al. Splice-switch oligonucleotide-based combinatorial platform prioritizes synthetic lethal targets CHK1 and BRD4 against MYC-driven hepatocellular carcinoma. Bioeng. Transl. Med. 2023, 8, e10363. [Google Scholar] [CrossRef]
- Liu, J.; Bhadra, M.; Sinnakannu, J.R.; Yue, W.L.; Tan, C.W.; Rigo, F.; Ong, S.T.; Roca, X. Overcoming imatinib resistance conferred by the BIM deletion polymorphism in chronic myeloid leukemia with splice-switching antisense oligonucleotides. Oncotarget 2017, 8, 77567–77585. [Google Scholar] [CrossRef]
- Mogilevsky, M.; Shimshon, O.; Kumar, S.; Mogilevsky, A.; Keshet, E.; Yavin, E.; Heyd, F.; Karni, R. Modulation of MKNK2 alternative splicing by splice-switching oligonucleotides as a novel approach for glioblastoma treatment. Nucleic Acids Res. 2018, 46, 11396–11404. [Google Scholar] [CrossRef]
- Tano, V.; Jans, D.A.; Bogoyevitch, M.A. Oligonucleotide-directed STAT3 alternative splicing switch drives anti-tumorigenic outcomes in MCF10 human breast cancer cells. Biochem. Biophys. Res. Commun. 2019, 513, 1076–1082. [Google Scholar] [CrossRef]
- Uehara, H.; Cho, Y.; Simonis, J.; Cahoon, J.; Archer, B.; Luo, L.; Das, S.K.; Singh, N.; Ambati, J.; Ambati, B.K. Dual suppression of hemangiogenesis and lymphangiogenesis by splice-shifting morpholinos targeting vascular endothelial growth factor receptor 2 (KDR). FASEB J. 2013, 27, 76–85. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef]
- Liang, X.H.; Nichols, J.G.; Hsu, C.W.; Vickers, T.A.; Crooke, S.T. mRNA levels can be reduced by antisense oligonucleotides via no-go decay pathway. Nucleic Acids Res. 2019, 47, 6900–6916. [Google Scholar] [CrossRef] [PubMed]
- Harigaya, Y.; Parker, R. No-go decay: A quality control mechanism for RNA in translation. Wiley Interdiscip Rev. RNA 2010, 1, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Denichenko, P.; Mogilevsky, M.; Clery, A.; Welte, T.; Biran, J.; Shimshon, O.; Barnabas, G.D.; Danan-Gotthold, M.; Kumar, S.; Yavin, E.; et al. Specific inhibition of splicing factor activity by decoy RNA oligonucleotides. Nat. Commun. 2019, 10, 1590. [Google Scholar] [CrossRef] [PubMed]
- Jayasinghe, R.G.; Cao, S.; Gao, Q.; Wendl, M.C.; Vo, N.S.; Reynolds, S.M.; Zhao, Y.; Climente-Gonzalez, H.; Chai, S.; Wang, F.; et al. Systematic Analysis of Splice-Site-Creating Mutations in Cancer. Cell Rep. 2018, 23, 270–281.e273. [Google Scholar] [CrossRef] [PubMed]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; The Cancer Genome Atlas Research Network; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8,705 Patients. Cancer Cell 2018, 34, 211–224.e216. [Google Scholar] [CrossRef]
- Seiler, M.; Peng, S.; Agrawal, A.A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P.G.; The Cancer Genome Atlas Research Network; Buonamici, S.; Yu, L. Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep. 2018, 23, 282–296.e284. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.X.; De Neef, E.; Thomas, J.D.; Sabio, E.; Rousseau, B.; Gigoux, M.; Knorr, D.A.; Greenbaum, B.; Elhanati, Y.; Hogg, S.J.; et al. Pharmacologic modulation of RNA splicing enhances anti-tumor immunity. Cell 2021, 184, 4032–4047.e4031. [Google Scholar] [CrossRef] [PubMed]
- North, K.; Benbarche, S.; Liu, B.; Pangallo, J.; Chen, S.; Stahl, M.; Bewersdorf, J.P.; Stanley, R.F.; Erickson, C.; Cho, H.; et al. Synthetic introns enable splicing factor mutation-dependent targeting of cancer cells. Nat. Biotechnol. 2022, 40, 1103–1113. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organ | Splicing Factor | Type of Alterations |
|---|---|---|
| Brain | SRSF1, SRSF3, HNRNPA1, HNRNPA2, HNRNPHK | Upregulation |
| Breast | SRSF1, SRSF3, SRSF4, SRSF5, SRSF6, TRA2B, HNRNPA1, HNRNPI RBM5, RBFOX2, HNRNPK SF3B1 | Upregulation Downregulation Somatic mutation |
| Bladder | SRSF1, SRSF3 | Upregulation |
| Colon | SRSF1, SRSF3, SRSF6, SRSF10, TRA2B HNRNPK, RBFOX2 | Upregulation Downregulation |
| Intestine | SRSF1 | Upregulation |
| Kidney | SRSF1, SRSF3 | Upregulation |
| Liver | SRSF3 | Upregulation |
| Lung | SRSF1, SRSF3, SRSF5, SRSF6, TRA2B RBM5, QKI RBM10, U2AF1 | Upregulation Downregulation Somatic mutation |
| Skin | SRSF3 HNRNPK SF3B1, SRSF2 | Upregulation Downregulation Somatic mutation |
| Thyroid | SRSF1, SRSF3 RBM10 | Upregulation Somatic mutation |
| Myeloid leukemias | SF3B1, SRSF2, U2AF1, ZRSR2 | Somatic mutation |
| Chronic lymphocytic leukemia | SF3B1 | Somatic mutation |
| Gene | Splice Variant | AS Event | Therapeutic Resistance | Cancer | Mechanism | Ref. |
|---|---|---|---|---|---|---|
| AR | AR-V7 | Cryptic exon usage | Androgen deprivation therapy | Prostate | Removes ligand-binding domain | [49] |
| BIM | Tyrosine kinase inhibitors | Lung | ||||
| BRACA1 | BRCA1-Δ11q | Alternative SS usage | PARP inhibitors | Breast, ovarian | Mutation removal | [50] |
| BARD1 | BARD1β | Exon 2,3 skipping | PARP inhibitors | Colon | Prevents BARD1/BRACA1 dimerization | [51] |
| BRAF | p61BRAF(V600E) | Exon 4-8 skipping | BRAF inhibitors | Thyroid, Skin | Inhibits downstream signaling | [52,53] |
| HER2 | ΔHER2 | Exon 16 skipping | mABs | Breast | Homodimer stabilization | [54] |
| MS4A1 | CD20-V1, -V2 | Aberrant 5’ UTR splicing | mABs | Lymphoma, Leukemia | Translation inhibition | [55] |
| TAK1 | TAK1ΔE12 | Exon 12 skipping | Chemotherapy | Breast | JNK/p38 activation | [56] |
| PKM | PKM2 | Isoform switch | Chemotherapy | Pancreatic | Unknown | [57] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonner, E.A.; Lee, S.C. Therapeutic Targeting of RNA Splicing in Cancer. Genes 2023, 14, 1378. https://doi.org/10.3390/genes14071378
Bonner EA, Lee SC. Therapeutic Targeting of RNA Splicing in Cancer. Genes. 2023; 14(7):1378. https://doi.org/10.3390/genes14071378
Chicago/Turabian StyleBonner, Elizabeth A., and Stanley C. Lee. 2023. "Therapeutic Targeting of RNA Splicing in Cancer" Genes 14, no. 7: 1378. https://doi.org/10.3390/genes14071378
APA StyleBonner, E. A., & Lee, S. C. (2023). Therapeutic Targeting of RNA Splicing in Cancer. Genes, 14(7), 1378. https://doi.org/10.3390/genes14071378